Ultra-Fast Liquid Chromatography vs. Spectrophotometry: A Strategic Guide for Pharmaceutical Analysis
This article provides a comprehensive comparison between Ultra-Fast Liquid Chromatography (UFLC) and Spectrophotometric methods for researchers and drug development professionals.
Ultra-Fast Liquid Chromatography vs. Spectrophotometry: A Strategic Guide for Pharmaceutical Analysis
Abstract
This article provides a comprehensive comparison between Ultra-Fast Liquid Chromatography (UFLC) and Spectrophotometric methods for researchers and drug development professionals. It explores the foundational principles of both techniques, detailing their specific applications from drug assay to impurity profiling. The content delivers practical troubleshooting guidance and outlines rigorous validation protocols tailored for complex matrices. Finally, it presents a strategic framework for method selection, empowering scientists to choose the optimal technique for accuracy, speed, and regulatory compliance in biomedical and clinical research.
Core Principles: Deconstructing UFLC and Spectrophotometry
Ultra-Fast Liquid Chromatography (UFLC) represents a significant technological evolution in chromatographic science, engineered specifically to achieve dramatic reductions in analysis time while maintaining or even enhancing chromatographic resolution and sensitivity. This advancement is particularly crucial in fields like pharmaceutical development, where the ability to rapidly analyze complex biological samples can significantly accelerate research and quality control processes. UFLC systems accomplish this primarily through the use of sub-2-micron particle columns and instrumentation capable of operating at significantly higher pressures (often exceeding conventional HPLC limits) compared to traditional High-Performance Liquid Chromatography (HPLC) [1]. The core principle involves optimizing the relationship between particle size, column length, operating pressure, and eluent velocity to achieve the highest possible plate count within a drastically reduced analysis time [2]. When applied to the discrimination between analytical techniques, such as comparing UFLC to spectrophotometric methods, UFLC's superior specificity and speed make it an powerful tool for complex analyses, such as pharmacokinetic studies where it can effectively separate and quantify a drug from its metabolites in a biological matrix [3].
Theoretical Foundations of Speed and Efficiency
The exceptional speed of UFLC is not the result of a single factor but the synergistic optimization of multiple chromatographic parameters. The fundamental goal is to achieve the highest efficiency, expressed as the number of theoretical plates (N), in the shortest possible analysis time, often represented by the column dead time (t₀) [2].
Optimization Schemes for Speed
Chromatographers employ different levels of optimization, each offering varying degrees of performance enhancement:
- One-Parameter Optimization: This basic approach involves selecting a column (fixed particle size and length) and then optimizing only the eluent velocity using the van Deemter equation to find the velocity that gives the minimal plate height. The limitation is that the analysis time is predetermined by the chosen column length, often resulting in sub-optimal performance [2].
- Two-Parameter Optimization: Here, the particle size is fixed, but both the column length and eluent velocity are optimized. Techniques like Poppe or kinetic plots are used, considering pressure and time constraints. This approach yields better performance than one-parameter optimization by calculating the ideal column length and velocity for a given analysis time [2].
- Three-Parameter Optimization: This is the most comprehensive scheme, simultaneously optimizing particle size, column length, and eluent velocity. This scenario, known as the Knox-Saleem limit, represents the absolute best possible separation performance. It dictates working at the van Deemter optimum velocity but with a specific combination of particle size and column length that maximizes plates for a given time [2].
The following table summarizes a comparison of these optimization schemes for a separation requiring a 4-second dead time, illustrating the performance gains from more comprehensive optimization strategies.
Table 1: Comparison of Optimization Schemes for a Separation with t₀ = 4 s
| Optimization Scheme | Particle Size (μm) | Column Length (mm) | Linear Velocity (cm/s) | Theoretical Plates (N) | Operating Pressure (bar) |
|---|---|---|---|---|---|
| One-Parameter | 1.8 (fixed) | 30 (fixed) | 0.75 | ~7,600 | 330 |
| Two-Parameter | 1.8 (fixed) | 53 | 1.33 | ~10,600 | 1,000 |
| Three-Parameter | 1.0 | 29 | 0.73 | ~14,900 | 1,000 |
Adapted from a comparison of optimization schemes for ultrafast separation [2].
The Role of System Pressure and Particle Size
The practical implementation of these theoretical optimizations relies on advanced engineering. The use of smaller particles (e.g., sub-2-μm) increases the resistance to flow, requiring higher operating pressures to achieve the optimal linear velocities. Modern UFLC systems are therefore designed to withstand pressures up to 1000-1500 bar, unlike traditional HPLC systems [2] [1]. This combination of high pressure and small particles creates a larger number of theoretical plates per unit time, enabling both rapid analysis and high resolution. The miniaturization of particles and the use of narrower-bore columns also contribute to lower solvent consumption, making the technique not only faster but also more cost-effective and environmentally friendly compared to methods using monolithic columns at high velocities [2].
Experimental Protocol: UFLC Method Development and Validation
This protocol outlines the development and validation of a UFLC method for the quantification of a small molecule drug (using Domperidone as an example) in human serum, culminating in its application to a pharmacokinetic study [3]. The workflow for this process is summarized in the following diagram.
Materials and Reagents
Table 2: Essential Research Reagents and Materials for UFLC Analysis of Domperidone in Serum
| Item | Specification / Example | Function / Purpose |
|---|---|---|
| UFLC System | Shimadzu UFLC with RF-10A XL fluorescence detector | Core instrumentation for ultra-fast separation and detection. |
| Analytical Column | Reversed-phase C18 column | Stationary phase for chromatographic separation of analytes. |
| Analyte Standard | Domperidone (DOM) | The target molecule for quantification. |
| Internal Standard (IS) | Propranolol Hydrochloride (PH) | Corrects for variability in sample preparation and injection. |
| Mobile Phase | 10 mM Phosphate Buffer (pH 3.1) and Methanol (62:38) | Liquid medium that carries the sample through the column. |
| Precipitation Solvent | Acetonitrile (ACN) | Removes proteins from the serum sample. |
| Serum Samples | Control human serum; study samples | The complex biological matrix containing the analyte. |
Based on the method for fluorescence detection of Domperidone [3].
Step-by-Step Procedure
Chromatographic Configuration
- Column: Install a reversed-phase C18 column and maintain at 40°C.
- Mobile Phase: Prepare a mixture of 10 mM phosphate buffer (pH adjusted to 3.1 with orthophosphoric acid) and methanol in a 62:38 (v/v) ratio. Filter and degas.
- UFLC Parameters: Set the flow rate to 1.0 mL/min and the injection volume to 20 μL.
- Detection: Configure the fluorescence detector with excitation at 282 nm and emission at 328 nm [3].
Sample Preparation Protocol
- Aliquot: Transfer 1 mL of human serum into a screw-capped tube.
- Spike Internal Standard: Add 100 μL of IS working solution (1500 ng/mL of Propranolol HCl) and vortex for 2 minutes.
- Protein Precipitation: Add 7 mL of acetonitrile, vortex for 2 minutes, and centrifuge at 6000 rpm for 15 minutes.
- Extract Processing: Transfer the organic supernatant layer and evaporate it to dryness under a vacuum.
- Reconstitution: Reconstitute the dried residue with 100 μL of mobile phase and inject 20 μL into the UFLC system [3].
Method Validation Tests
The developed method must be rigorously validated against standard criteria to ensure reliability for pharmacokinetic studies. The key parameters and their acceptance criteria, as demonstrated in the domperidone study, are summarized below.
Table 3: Method Validation Parameters and Results for Domperidone UFLC Assay
| Validation Parameter | Description & Procedure | Acceptance Criteria / Result |
|---|---|---|
| Calibration Curve | Analyze standards across concentration range (e.g., 10 - 10,000 ng/mL). | Linear relationship with R² > 0.99 [3]. |
| Precision (Intra-day & Inter-day) | Analyze QC samples (15, 4750, 9500 ng/mL) in replicates (n=5) over different days. | Coefficient of Variation (CV) < 5% [3]. |
| Accuracy | Compare measured concentration of QC samples to known true value. | Relative Error (RE) < 5% [3]. |
| Low Limit of Quantification (LLOQ) | Determine the lowest standard that can be measured with acceptable precision and accuracy. | CV and RE < 20%; established at 15 ng/mL for DOM [3]. |
| Recovery | Compare analyte peak area from extracted samples to non-extracted standards. | Mean recovery > 96% [3]. |
| Stability | Evaluate bench-top, freeze-thaw, and long-term storage stability of analyte in serum. | Concentration change within ±15% of nominal [3]. |
| Robustness | Deliberately vary method parameters (flow rate ±0.2 mL/min, pH ±0.2, temp ±5°C). | Method performance remains within acceptance criteria [3]. |
Application in Pharmacokinetic Study and Technique Discrimination
The validated UFLC method was successfully applied to a pharmacokinetic study comparing different dosage forms of domperidone in healthy human volunteers. The study demonstrated the practical utility of UFLC's speed and specificity for analyzing time-sensitive biological samples [3].
Pharmacokinetic Results
The key pharmacokinetic parameters derived from the UFLC analysis highlight its ability to discriminate between different drug release profiles, a task where spectrophotometric methods may lack specificity:
- Immediate-Release (IR) Buccal Patch: Cmax = 129.7 ng/mL, Tmax = 1.5 h, AUC0–24 = 455.1 ng·h/mL
- Controlled-Release (CR) Buccal Patch: Cmax = 145.7 ng/mL, Tmax = 5.25 h, AUC0–24 = 911.0 ng·h/mL [3]
The clear discrimination in Tmax and the shape of the concentration-time curve for the CR formulation, as determined by UFLC, provides definitive evidence of modified release, which might be challenging to deconvolute using non-separative spectrophotometric techniques, especially if metabolites are present.
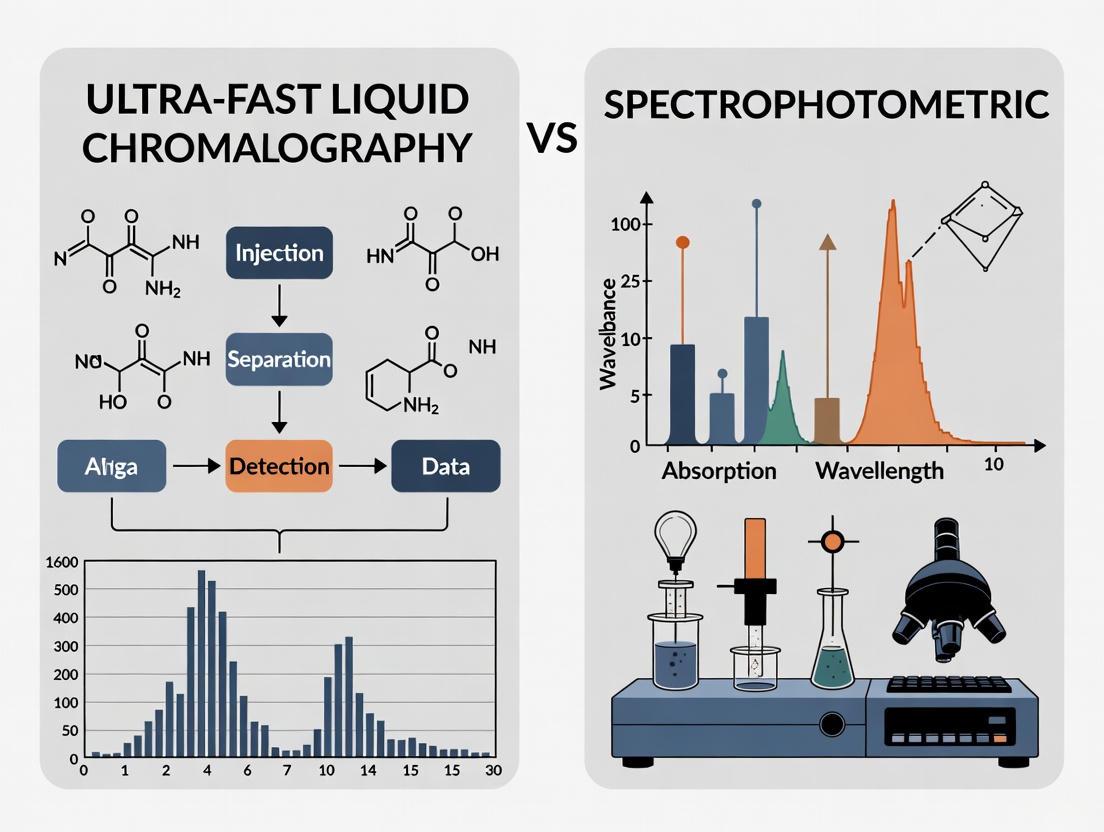
UFLC vs. Spectrophotometry: A Comparative Perspective
In the context of the broader thesis on technique discrimination, the application note demonstrates critical distinctions. While UV-Vis spectrophotometry is often cheaper and simpler for dissolution testing, UFLC offers superior specificity by physically separating the API from degradation products or metabolites prior to detection [2] [3]. This is critical in complex biological matrices like serum, where numerous interfering compounds are present. Furthermore, UFLC provides a wider linear dynamic range, making it more versatile for early drug development when different formulations and strengths are screened. The primary advantage of traditional UV has been speed, but as this protocol shows, UFLC closes this gap dramatically, achieving run times of around 6-8 minutes for domperidone, making it competitive for high-throughput analyses like pharmacokinetic studies [2] [3].
The Beer-Lambert Law (also known as Beer's Law) is a fundamental principle in optical spectroscopy that describes the quantitative relationship between the absorption of light and the properties of the material through which the light is traveling [4] [5]. This law enables researchers to make precise measurements of substance concentration and purity by analyzing how materials absorb light at specific wavelengths, forming the cornerstone of many analytical techniques used in pharmaceutical research, quality control, and method discrimination studies [6].
In the context of ultra-fast liquid chromatography versus spectrophotometric method discrimination research, understanding the Beer-Lambert Law is crucial for evaluating the complementary strengths and limitations of these analytical techniques. While chromatographic methods separate compounds, spectrophotometric methods relying on Beer-Lambert principles provide rapid, non-destructive quantification essential for method validation and comparative analysis [7] [8].
Theoretical Foundation
Fundamental Principles
The Beer-Lambert Law establishes that when a beam of monochromatic light passes through a solution containing an absorbing substance, the attenuation of light is directly proportional to the concentration of the absorbing species and the path length the light travels through the solution [9] [6]. The law is mathematically expressed as:
A = εlc
Where:
- A is the absorbance (dimensionless)
- ε is the molar absorptivity or molar extinction coefficient (L·mol⁻¹·cm⁻¹)
- l is the path length through the sample (cm)
- c is the concentration of the absorbing species (mol/L) [4] [9]
The absorbance has a logarithmic relationship to the transmittance, which is defined as the ratio of transmitted intensity (I) to incident intensity (I₀) [4]:
A = log₁₀(I₀/I)
This relationship means that each unit increase in absorbance corresponds to a tenfold decrease in transmittance [4].
Historical Context
The development of what is now known as the Beer-Lambert Law spans nearly two centuries of scientific discovery. French scientist Pierre Bouguer first documented the exponential attenuation of light in the atmosphere in 1729 [5] [10]. Johann Heinrich Lambert later formalized this mathematical relationship in his 1760 work "Photometria," establishing that light intensity decreases exponentially with path length through an absorbing medium [5] [10].
In 1852, August Beer extended these principles to colored solutions, demonstrating that absorbance is proportional to concentration in addition to path length [5] [10]. The modern formulation combining both relationships was first presented by Robert Luther and Andreas Nikolopulos in 1913 [5]. This historical evolution explains why the law is sometimes referenced with varying combinations of the three scientists' names (Bouguer-Beer-Lambert Law) [10].
Visualizing the Beer-Lambert Law
The following diagram illustrates the fundamental components and relationships described by the Beer-Lambert Law:
Beer-Lambert Law Components Diagram: Visualization of the key elements in spectrophotometric measurements based on Beer-Lambert principles.
Quantitative Relationships
Absorbance and Transmittance Correlation
The Beer-Lambert Law establishes an inverse logarithmic relationship between absorbance and transmittance. This relationship means that as absorbance increases, transmittance decreases exponentially [4]. The following table illustrates this fundamental correlation:
Table 1: Absorbance and Transmittance Values
| Absorbance (A) | Transmittance (T) | Transmitted Light (%) |
|---|---|---|
| 0 | 1 | 100% |
| 0.5 | 0.316 | 31.6% |
| 1 | 0.1 | 10% |
| 2 | 0.01 | 1% |
| 3 | 0.001 | 0.1% |
| 4 | 0.0001 | 0.01% |
| 5 | 0.00001 | 0.001% |
Source: Adapted from Edinst resource on Beer-Lambert Law [4]
This quantitative relationship enables researchers to interpret spectrophotometric data accurately and understand how minute changes in concentration or path length affect light transmission through samples.
Molar Absorptivity and Its Significance
The molar absorptivity (ε), also known as the molar extinction coefficient, is a fundamental property of each chemical species that indicates how strongly a compound absorbs light at a specific wavelength [9] [6]. This parameter is intrinsic to each molecule and depends on factors such as:
- Molecular structure and electronic transitions
- Solvent composition and properties
- Wavelength of incident light
- Temperature and pH conditions [6]
Compounds with high molar absorptivity values are more easily detected at low concentrations, making this parameter crucial for method sensitivity assessment in pharmaceutical analysis [9].
Practical Applications in Pharmaceutical Analysis
Concentration Determination in Drug Formulations
The primary application of Beer-Lambert Law in pharmaceutical research is the quantification of active pharmaceutical ingredients (APIs) in formulations. The linear relationship between absorbance and concentration enables the creation of calibration curves for accurate determination of unknown concentrations [4] [7].
In a comparative study of HPLC and UV spectrophotometric methods for determination of favipiravir, both techniques demonstrated excellent linearity with correlation coefficients greater than 0.999 within a concentration range of 10-60 μg/mL [7]. The spectrophotometric method provided accuracy within 99.83-100.45%, making it a reliable technique for quality control of this antiviral medication [7].
Method Comparison and Validation
The Beer-Lambert Law provides the theoretical foundation for validating spectrophotometric methods against separation techniques like ultra-fast liquid chromatography. Key validation parameters include:
- Linearity: Verification of the A = εlc relationship across concentration ranges
- Specificity: Confirmation that absorbance measurements are free from interference
- Precision: Evaluation of intra-day and inter-day reproducibility
- Accuracy: Determination of percentage recovery of known concentrations [7]
Research comparing UPLC and HPLC methods for vitamin C determination demonstrated that both techniques could be optimized for specific analytical needs, with spectrophotometric methods offering advantages in speed and simplicity for certain applications [8].
Experimental Protocols
Protocol 1: UV Spectrophotometric Determination of Favipiravir
This protocol outlines the methodology for quantifying favipiravir in pharmaceutical formulations using UV spectrophotometry based on Beer-Lambert principles [7].
Research Reagent Solutions
Table 2: Essential Materials for Favipiravir Analysis
| Reagent/Material | Specifications | Function |
|---|---|---|
| Favipiravir standard | Pharmaceutical grade | Reference standard for calibration |
| Deionized water | Milli-Q purified | Solvent for standard and sample preparation |
| UV spectrophotometer | Double-beam with 1.0 cm quartz cells | Absorbance measurement at 227 nm |
| Analytical balance | MettlerToledo, 0.1 mg precision | Accurate weighing of standards and samples |
| Volumetric flasks | Class A, various sizes | Precise solution preparation |
| Filter paper | Whatman No. 42 | Sample clarification |
Procedure
Standard Solution Preparation: Prepare a stock standard solution of favipiravir (1000 μg/mL) in deionized water. Sonicate and filter through a 0.22 μm filter.
Calibration Standards: Dilute the stock solution with deionized water to obtain standard solutions in the concentration range of 10-60 μg/mL.
Sample Preparation: Weigh and finely powder ten favipiravir tablets (200 mg). Transfer tablet powder equivalent to 50 mg of favipiravir to a 50 mL volumetric flask and dissolve in 30 mL deionized water. Shake for 30 minutes, then dilute to volume with deionized water to obtain 1000 μg/mL concentration. Filter using Whatman No. 42 filter paper.
Wavelength Determination: Scan the favipiravir solution (30 μg/mL) between 200-800 nm using deionized water as blank. Identify maximum absorption at 227 nm.
Absorbance Measurement: Measure absorbance of all standard and sample solutions at 227 nm using 1.0 cm quartz cells with deionized water as reference.
Calibration Curve: Plot absorbance versus concentration of standard solutions and determine the regression equation.
Concentration Calculation: Calculate the concentration of favipiravir in sample solutions using the regression equation.
Data Analysis
The following workflow diagram illustrates the experimental process for spectrophotometric drug analysis:
Spectrophotometric Analysis Workflow: Step-by-step procedure for drug quantification using Beer-Lambert Law.
Protocol 2: Comparative Method Validation Using Chromatography and Spectrophotometry
This protocol outlines the procedure for comparing ultra-fast liquid chromatography with spectrophotometric methods for pharmaceutical analysis [7] [8].
Materials and Equipment
- Ultra-performance liquid chromatography system with UV detector
- UV-Visible spectrophotometer with double beam and 1.0 cm quartz cells
- Chromatographic column: C18 column (e.g., Inertsil ODS-3, 4.6 × 250 mm, 5 μm)
- Mobile phase: Appropriate mixture based on analyte (e.g., sodium acetate buffer pH 3.0:acetonitrile, 85:15 v/v for favipiravir)
- Standard solutions of analyte across validation range
Procedure
System Preparation: Equilibrate both UPLC and spectrophotometry systems according to manufacturer specifications.
Method Development: Optimize chromatographic conditions (mobile phase composition, flow rate, column temperature) and spectrophotometric parameters (wavelength selection, bandwidth).
Linearity Assessment: Analyze standard solutions across the concentration range (e.g., 10-60 μg/mL) using both techniques. Perform triplicate measurements.
Precision Evaluation: Determine intra-day precision by analyzing six replicates of quality control samples at low, medium, and high concentrations within the same day. Assess inter-day precision over three consecutive days.
Accuracy Testing: Perform recovery studies by spiking placebo with known amounts of analyte at three concentration levels. Calculate percentage recovery.
Specificity Verification: Analyze placebo formulation and check for interference at retention time (chromatography) or wavelength (spectrophotometry).
Data Comparison: Statistically compare results from both methods using appropriate tests (e.g., ANOVA, regression analysis).
Advanced Applications and Current Research
Integration with Multivariate Analysis
Modern applications of Beer-Lambert principles extend beyond simple single-wavelength measurements. Fourier-transform infrared (FTIR) spectroscopy combined with chemometric analysis enables discrimination of complex biological samples based on their absorption fingerprints [11].
In nectar discrimination studies, FTIR spectroscopy successfully differentiated samples from different plant species and geographical origins by analyzing specific spectral regions [11]:
- Carbohydrate fingerprint region (1200-950 cm⁻¹)
- C-H stretching zone (2935-2885 cm⁻¹)
This approach demonstrates how Beer-Lambert principles underpin advanced spectroscopic techniques for sample classification and authentication in complex matrices.
Method Discrimination Research
The comparative evaluation of analytical techniques represents a significant research area where Beer-Lambert Law provides the fundamental framework for assessing spectrophotometric method performance against separation techniques [7] [8].
Table 3: Comparison of Spectrophotometric and Chromatographic Methods
| Parameter | UV Spectrophotometry | Ultra-Fast Liquid Chromatography |
|---|---|---|
| Principle | Beer-Lambert Law (light absorption) | Partitioning between stationary and mobile phases |
| Analysis Time | Rapid (minutes) | Moderate to fast (5-15 minutes) |
| Sensitivity | Good for strong chromophores | Excellent with various detection options |
| Selectivity | Limited for mixtures | High (separation of components) |
| Sample Preparation | Minimal to moderate | Often requires extensive preparation |
| Cost | Lower equipment and operational costs | Higher initial investment and running costs |
| Applications | Quantitative analysis of single components | Complex mixtures, impurity profiling |
In favipiravir analysis research, both spectrophotometric and liquid chromatographic methods demonstrated excellent linearity (r > 0.999) and precision (RSD < 2%), with each technique offering distinct advantages for specific application scenarios [7]. The spectrophotometric method provided simplicity and rapid analysis, while chromatography offered superior selectivity for complex matrices.
Limitations and Considerations
Fundamental Limitations
Despite its widespread utility, the Beer-Lambert Law has specific limitations that researchers must consider when developing analytical methods:
Concentration Limitations: Deviations from linearity occur at high concentrations (>0.01 M) due to molecular interactions and changes in refractive index [6] [12]
Chemical Factors: Associations between solute molecules, equilibrium processes, and pH-dependent speciation can affect absorbance-concentration linearity [6]
Optical Considerations: Scattering, fluorescence, and stray light can lead to inaccurate absorbance measurements [10]
Electromagnetic Effects: The fundamental wave nature of light creates situations where the Beer-Lambert Law provides only an approximation, particularly in strongly absorbing media or at interfaces between materials with different refractive indices [10]
Practical Considerations for Method Development
When implementing Beer-Lambert Law for quantitative analysis, several practical aspects require attention:
Wavelength Selection: Optimal analysis occurs at the wavelength of maximum absorption (λmax) where the molar absorptivity is highest and the method is most sensitive [7]
Blank Preparation: The reference solution should match the sample matrix as closely as possible to compensate for solvent and matrix effects
Path Length Consistency: Using matched cuvettes with identical path lengths ensures accurate absorbance measurements [4]
Concentration Range: Maintaining analyte concentration within the linear range of the instrument is essential for accurate quantification [6]
The Beer-Lambert Law remains a cornerstone of modern analytical spectroscopy, providing the fundamental relationship between light absorption and material properties that enables quantitative analysis across pharmaceutical, environmental, and biological applications. In the context of ultra-fast liquid chromatography versus spectrophotometric method discrimination research, understanding the principles, capabilities, and limitations of Beer-Lambert-based methods is essential for selecting appropriate analytical techniques for specific applications.
While chromatographic methods offer superior separation capabilities for complex mixtures, spectrophotometric methods based on Beer-Lambert principles provide rapid, cost-effective quantification for single-component analysis and quality control applications. The continued development of spectroscopic technologies, including integration with multivariate analysis and advanced detection systems, ensures that the Beer-Lambert Law will maintain its relevance as a fundamental principle in analytical science.
In the modern analytical laboratory, the synergy between separation science and detection technology forms the cornerstone of effective research and development. This application note provides a detailed exploration of two pivotal instrumental domains: Ultra-Fast Liquid Chromatography (UFLC) systems, with a focus on pump and column technologies that enable rapid separations, and spectrophotometric detection systems, particularly the lamps that are fundamental to their operation. Framed within broader research on ultrafast liquid chromatography versus spectrophotometric method discrimination, this document offers structured quantitative data, detailed experimental protocols, and visual workflows to support scientists in drug development and related fields. The drive for increased throughput without compromising data integrity has made understanding these instrumental fundamentals more critical than ever [13].
Ultrafast Liquid Chromatography (UFLC) Instrumentation
Core Principles and Theoretical Foundation
Ultrafast Liquid Chromatography achieves dramatic reductions in analysis time—from tens of minutes to under a minute—while maintaining, or even enhancing, chromatographic resolution. This is principally governed by the van Deemter equation, which describes the relationship between linear velocity (flow rate) and column efficiency, expressed as Height Equivalent to a Theoretical Plate (HETP) [14] [13]. The equation is represented as:
H = A + B/μ + Cμ
Where H is the HETP, μ is the linear velocity, and A, B, and C are coefficients for eddy diffusion, longitudinal diffusion, and resistance to mass transfer, respectively. The key to UFLC lies in minimizing the A and C terms, which is achieved by using smaller, uniformly sized particles for column packing. This results in a "flatter" van Deemter curve, allowing operation at higher linear velocities without a significant loss of efficiency [14] [13]. The fundamental relationship between particle size and pressure drop cannot be overlooked, as pressure increases with the inverse square of the particle diameter, making hardware capable of withstanding very high pressures (e.g., >15,000 psi) a prerequisite for exploiting sub-2 μm particles [14] [15].
Critical UHPLC Pump Technologies
The pump is the heart of any UHPLC system, and for ultrafast applications, its requirements are stringent. Modern UHPLC pumps must deliver precise, pulse-free flow at pressures up to 19,000 psi (approximately 1300 bar) [15]. Advanced designs, such as the asymmetric twin-piston solvent delivery system with Slow Suction, Quick Delivery (SSQD) technology, provide significantly better flow and pressure characteristics than conventional reciprocating designs. This high stability is particularly crucial for detectors sensitive to flow pulsation, such as mass spectrometers, refractive index (RI), and electrochemical detectors [16]. Furthermore, to accommodate the narrow peaks produced by UFLC (which can be just a few seconds wide), the pump must be integrated into a system with a minimal dwell volume (the volume between the point of mixing and the column head). A low dwell volume ensures rapid gradient formation and sharper separations, which is vital for fast cycle times [14].
Key Components for UFLC
Table 1: Essential UHPLC System Components for Ultrafast Separations
| Component | Key Specification | Impact on UFLC Performance | Typical Vendor Examples |
|---|---|---|---|
| Pump | Pressure rating (up to 19,000 psi), flow precision, low pulsation | Enables use of sub-2 μm particles at optimal flow rates; ensures mobile phase stability [16] [15]. | Agilent InfinityLab, JASCO PU-4280/85 [17] [16] |
| Autosampler | Fast injection cycle (e.g., 10s), low carryover, thermostatted | Reduces total cycle time; maintains sample integrity [13]. | Integrated modules in Agilent, JASCO, Shimadzu systems [17] [13] |
| Column Oven | Forced-air circulation, rapid heating/cooling, precise temperature control | Essential for elevated temperature LC; eliminates temperature gradients that distort peaks [13]. | JASCO CO-4065, Agilent InfinityLab [17] [16] |
| Detector | High acquisition rate (>5 Hz), low-volume flow cell | Captures narrow peaks with sufficient data points (10-15 points/peak) for accurate quantification [15]. | JASCO FP-4020, RI-4035 [16] |
| Column | Sub-2 μm or 2-3 μm particles; narrow bore (e.g., 2.1 mm i.d.); stable chemistry | Provides high efficiency; reduces solvent consumption; increases sensitivity [14] [13] [15]. | Various C18, HILIC, and polar-embedded phases [15] |
Spectrophotometric Detection and Lamp Technologies
In the context of chromatography, spectrophotometric detection—primarily UV-Vis—is a workhorse for quantifying analytes as they elute from the column. The light source, typically a deuterium lamp for the UV range and a tungsten-halogen lamp for the visible range, is fundamental to this process. These lamps generate a broad spectrum of light, which is then passed through a monochromator to select specific wavelengths for probing the sample in the flow cell [16]. The stability and intensity of the lamp directly impact the sensitivity, signal-to-noise ratio, and baseline drift of the chromatographic output. As a pioneer in optical spectroscopy, manufacturers like JASCO incorporate advanced optical designs that are critical for detectors with unrivaled performance, including fluorescence and circular dichroism detectors [16].
Beyond Chromatography: Spectrophotometry for Sample Discrimination
Standalone spectrophotometers play a vital role in research for material characterization and discrimination. They function by quantifying how much light a sample absorbs or transmits across a range of wavelengths, providing a unique "fingerprint." This is crucial in applications like raw material identification and quality control of colored products. HunterLab emphasizes that spectrophotometry neutralizes the effects of visual discrimination by controlling variables such as lighting, viewing angle, and surface texture, which can drastically alter human color perception [18]. This provides a quantifiable and repeatable measurement, ensuring consistency in industries from pharmaceuticals to plastics.
Experimental Protocols
Protocol 1: Method Transfer from HPLC to UHPLC for Impurity Profiling
Objective: To adapt an existing HPLC impurity method to a faster UHPLC method while maintaining or improving chromatographic resolution.
Materials:
- Original Method: HPLC system, 150 mm x 4.6 mm, 5 μm C18 column.
- Target Method: UHPLC system (pressure capability ≥ 15,000 psi), 50 mm x 2.1 mm, 1.8 μm C18 column of the same brand and chemistry.
- Samples: Drug substance and impurity mixture in appropriate diluent [15].
Procedure:
- Geometric Scaling: Use a method scaling calculator (often provided by column vendors) to transfer the gradient. The scaling factor is calculated based on the change in column volume (L × d²). For the example above, the factor is (50/150) × (2.1²/4.6²) ≈ 0.07. Multiply all gradient time segments (e.g., hold, ramp, equilibration) by this factor [15].
- Flow Rate Adjustment: Scale the flow rate proportionally to the cross-sectional area of the column. The factor is (d₂² / d₁²) = (2.1² / 4.6²) ≈ 0.21. Multiply the original flow rate by this factor.
- Injection Volume: Scale the injection volume by the same factor used for the gradient (0.07) to maintain mass load and detection sensitivity.
- Detector Settings: Adjust the detector data acquisition rate to 5-10 Hz to ensure at least 10-15 data points are collected across the now-narrower peaks [15].
- Method Execution and Verification: Run the scaled method. Compare the chromatographic profile, specifically the resolution between critical pairs (e.g., API and closest eluting impurity), with the original HPLC method. Fine-tune the gradient or temperature if necessary to achieve equivalent or superior resolution [15].
Protocol 2: Ultrafast Analysis of Xanthine Derivatives
Objective: To separate four xanthine derivatives in under 1.5 minutes using an optimized UFLC system.
Materials:
- Instrumentation: UFLC system (e.g., Shimadzu Prominence UFLC) with low-dispersion tubing [13].
- Column: Shim-pack XR-ODS (50 mm x 3.0 mm, 2.2 μm) or equivalent [13].
- Mobile Phase: A suitable water-acetonitrile gradient.
- Standards: Xanthine, theobromine, theophylline, caffeine (10 μg/mL each) [13].
Procedure:
- System Configuration: Ensure the system is configured for ultrafast work, with minimal internal volume capillaries and a fast-response detector.
- Chromatographic Conditions:
- Flow Rate: 2.0 mL/min
- Column Temperature: 60°C
- Injection Volume: 10 μL
- Detection: UV absorbance at 210 nm
- Gradient: Program a rapid gradient (e.g., from 5% to 50% B in 1 minute) tailored to the analyte properties [13].
- Analysis: Inject the standard mixture. The total run time, including column equilibration, should be approximately 1.2 minutes, yielding a baseline separation of all four compounds [13].
Integrated Workflow and Data Analysis
The successful implementation of UFLC and spectrophotometric discrimination requires a coherent strategy that integrates instrument selection, method development, and data processing. The following diagram illustrates the logical decision pathway for developing an ultrafast chromatographic method.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for UFLC and Spectrophotometric Research
| Item | Function / Application | Specific Example / Note |
|---|---|---|
| Sub-2 µm Analytical Columns | High-efficiency core-shell or fully porous particles for fast separations [14] [15]. | Various C18, C8, phenyl, and HILIC chemistries from major suppliers. |
| UHPLC-Grade Solvents | Low UV cutoff and minimal particulate matter to prevent baseline noise and column clogging [17]. | Acetonitrile, methanol, and water with 0.1% formic acid or ammonium formate [19]. |
| Deuterium & Tungsten Lamps | Stable light source for UV-Vis detection; critical for sensitivity and long-term baseline stability [16]. | Standard components in spectrophotometers and HPLC-UV detectors. |
| Polar-Embedded Phase Columns | Provides alternative selectivity for challenging separations of polar compounds [15]. | Used in method screening protocols to find optimal starting conditions. |
| MS-Grade Additives | Volatile buffers for mass spectrometric detection to avoid ion source contamination [19]. | Formic acid, ammonium formate, ammonium hydroxide. |
| On-line Filter/Frit | Protects the analytical column from particulates, extending column life [14]. | Placed between injector and column; requires regular maintenance. |
The push for greater analytical throughput is unequivocally linked to advances in instrumental design, particularly in the realms of UHPLC and spectrophotometry. This deep dive underscores that achieving ultrafast separations is a systematic process relying on the synergistic combination of robust high-pressure pumps, columns packed with small particles, and detectors with fast response times. Simultaneously, the role of spectrophotometric lamps as stable, reproducible light sources is fundamental to both integrated detection and standalone material discrimination. By applying the detailed protocols, workflows, and component knowledge outlined in this application note, researchers and drug development professionals can significantly enhance their operational efficiency and data quality.
Inherent Strengths and Limitations of Each Technique
In the modern pharmaceutical laboratory, the choice of analytical technique is pivotal to the success of drug development and quality control. Two methodologies frequently at the forefront of this decision are Ultra-Fast Liquid Chromatography (UFLC) and spectrophotometric methods. The former represents the evolution of high-performance liquid chromatography (HPLC) into a faster, more efficient format leveraging sub-2μm particles and higher pressure systems [20]. The latter, a mainstay of analytical chemistry, has experienced a resurgence through coupling with advanced chemometrics [21]. This application note provides a structured comparison of these techniques, offering detailed protocols to guide researchers in method selection and implementation within drug development workflows.
Technical Comparison: UFLC vs. Spectrophotometry
The following table summarizes the core characteristics, strengths, and limitations of UFLC and modern spectrophotometry for pharmaceutical analysis.
Table 1: Core characteristics and performance comparison of UFLC and Spectrophotometry.
| Feature | Ultra-Fast Liquid Chromatography (UFLC) | Spectrophotometry |
|---|---|---|
| Basic Principle | Separation of components using a pressurized liquid mobile phase and a stationary phase, followed by detection [20]. | Measurement of light absorption by molecules in a solution at specific wavelengths [21]. |
| Key Instrumentation | UHPLC pumps (>400 bar), sub-2μm particle columns, autosampler, DAD/UV/FL/MS detectors [20]. | UV-Vis spectrophotometer, light source (deuterium/tungsten), monochromator, sample cuvette, photodiode array detector [21]. |
| Primary Strength | High selectivity and specificity; can resolve complex mixtures accurately [22]. | Simplicity, speed of analysis, low operational cost, and ease of use [22]. |
| Primary Limitation | Higher instrumentation and maintenance costs; requires skilled operation [22] [20]. | Low selectivity in complex matrices; susceptible to interference from excipients and impurities [22]. |
| Separation Capability | Excellent; physically separates analytes from impurities and matrix components [20]. | None; measures total absorption of the sample without physical separation [22]. |
| Sensitivity | High (e.g., ng/mL levels with MS detection) [23]. | Moderate to good; limited by the analyte's molar absorptivity [22]. |
| Analysis Speed | Very fast (1-5 minutes per sample with modern UHPLC) [20]. | Extremely fast (seconds to minutes per sample) [21]. |
| Sample Consumption | Low (1-10 μL typical for UHPLC) [20]. | Moderate to high (often requires mL volumes for standard cuvettes) [22]. |
| Greenness (AGREE Metric) | Lower due to higher solvent consumption and energy use [22]. | Higher due to minimal solvent use and lower energy requirements [22]. |
Experimental Protocols
Protocol for UFLC-DAD Analysis of an Active Pharmaceutical Ingredient (API)
This protocol, adapted from a validation study for Metoprolol Tartrate (MET), outlines the steps for quantifying an API in a tablet formulation using UFLC-DAD [22].
1. Research Reagent Solutions: Table 2: Essential reagents and materials for the UFLC-DAD protocol.
| Item | Specification / Function |
|---|---|
| UFLC-DAD System | System capable of pressures up to 1000 bar, with a Diode Array Detector (DAD). |
| Analytical Column | Reversed-phase C18 column (e.g., 100 mm x 2.1 mm, 1.7-1.8 μm particle size). |
| API Reference Standard | High-purity (>98%) standard for calibration. |
| Mobile Phase A | Aqueous phase (e.g., 0.1% Formic Acid in Ultrapure Water). |
| Mobile Phase B | Organic phase (e.g., Acetonitrile or Methanol). |
| Ultrapure Water (UPW) | Solvent for preparation of standards and samples. |
2. Sample Preparation:
- Crush not less than 20 tablets into a fine, homogeneous powder.
- Accurately weigh a portion of the powder equivalent to the API content of one tablet.
- Transfer the powder to a volumetric flask and dissolve in UPW with the aid of sonication for 10-15 minutes.
- Dilute to volume with UPW and mix well.
- Filter the solution through a 0.22 μm or 0.45 μm membrane filter before injection into the UFLC system.
3. Instrumental Parameters:
- Column Temperature: 30 - 40 °C
- Injection Volume: 1 - 5 μL
- Flow Rate: 0.3 - 0.5 mL/min
- Mobile Phase Gradient:
- Time = 0 min: 5% B
- Time = 3.0 min: 95% B
- Time = 3.5 min: 95% B
- Time = 3.6 min: 5% B
- Time = 5.0 min: 5% B (equilibration)
- DAD Detection: Wavelength optimized for the API (e.g., 223 nm for MET).
4. Data Analysis:
- Inject a series of standard solutions to construct a calibration curve (e.g., 1-100 μg/mL).
- Inject the prepared sample solutions.
- Quantify the API concentration in the sample by comparing the peak area to the calibration curve.
Diagram 1: UFLC-DAD analysis workflow.
Protocol for Chemometrics-Assisted Spectrophotometric Analysis of an API
This protocol uses a full-spectrum approach with chemometrics to overcome the selectivity limitations of traditional spectrophotometry, ideal for quality control checks [21] [22].
1. Research Reagent Solutions: Table 3: Essential reagents and materials for the spectrophotometric protocol.
| Item | Specification / Function |
|---|---|
| UV-Vis Spectrophotometer | Instrument with a photodiode array detector capable of recording full spectra (200-800 nm). |
| Software | Chemometrics software for data analysis (e.g., for PCR or PLS regression). |
| API Reference Standard | High-purity standard for calibration. |
| Cuvettes | Quartz or UV-transparent plastic for spectral measurements. |
| Ultrapure Water (UPW) | Solvent for preparation of standards and samples. |
2. Sample Preparation:
- Follow the same tablet powdering and weighing procedure as in the UFLC protocol (Steps 1-3).
- Transfer the powder to a volumetric flask and dissolve in UPW with sonication.
- Dilute to volume and mix well. No filtration is strictly necessary if the solution is clear.
3. Spectral Acquisition:
- Fill a cuvette with UPW to collect a blank baseline spectrum.
- Prepare a series of standard solutions covering the expected concentration range of the API.
- Record the full UV-Vis spectrum (e.g., 200-400 nm) for each standard and the sample solution.
4. Chemometric Analysis & Quantification:
- Spectral Preprocessing: Apply techniques like Standard Normal Variate (SNV) or derivatives to reduce baseline drift and light scattering effects.
- Model Development: Use the standard spectra to build a multivariate calibration model, such as Partial Least Squares (PLS) regression. This model correlates the spectral data to the known concentrations.
- Prediction: Use the developed PLS model to predict the API concentration in the unknown sample solutions based on their spectra.
Diagram 2: Spectrophotometric analysis workflow.
Application in Drug Development: A Case Study
A recent study directly compared a UFLC-DAD method and a traditional spectrophotometric method for quantifying Metoprolol Tartrate (MET) in tablets [22]. The study provided a clear illustration of the strengths and limitations of each technique in a practical context.
UFLC-DAD Performance: The UFLC method demonstrated superior selectivity by successfully separating MET from tablet excipients and any potential degradation products. It showed excellent linearity, accuracy, and precision, and was successfully applied to tablets with two different dosage strengths (50 mg and 100 mg). Its robustness against interferences is its primary advantage for rigorous quality control.
Spectrophotometric Performance: The spectrophotometric method was simpler, faster, and more cost-effective. It also showed good precision and accuracy for the 50 mg tablets. However, it reached its limit for the 100 mg tablets due to the need for sample dilution to remain within the linear range of the Beer-Lambert law, highlighting a key limitation in its dynamic range compared to UFLC.
Greenness Assessment: Using the Analytical GREEnness (AGREE) metric, the study conclusively showed that the spectrophotometric method had a significantly higher greenness score, making it the more environmentally friendly choice due to its minimal solvent consumption [22].
The choice between UFLC and spectrophotometry is not a matter of one technique being universally superior, but rather of selecting the right tool for the specific analytical question and context.
UFLC is the undisputed choice when the sample matrix is complex, the target analyte is at a low concentration, or when specificity against interferences and degradation products is paramount. Its application is critical in discovery chemistry, pharmacokinetic studies, and rigorous stability testing [23] [24].
Spectrophotometry, especially when enhanced with chemometrics, offers a powerful and efficient alternative for high-throughput routine analysis, especially in quality control environments where cost, speed, and environmental impact are significant factors, and where the matrix is well-understood and relatively simple [21] [22].
Researchers and drug development professionals are advised to use this structured comparison and the accompanying protocols to make an informed decision that balances the need for analytical rigor with the practical constraints of efficiency and cost.
Spectral Interference and Matrix Effects in Spectrophotometry
Spectral interference and matrix effects are fundamental phenomena in analytical chemistry that can significantly compromise the accuracy and reliability of quantitative measurements. Within the broader context of ultra-fast liquid chromatography (UFLC) versus spectrophotometric method discrimination research, understanding these effects is paramount for developing robust, high-throughput analytical methods. Spectrophotometric techniques, including atomic absorption and X-ray fluorescence, are highly susceptible to these interferences, which manifest as inaccurately high or low concentration readings due to overlapping signals or sample matrix components. This application note provides a detailed examination of these interference types, presents systematic correction methodologies, and establishes experimental protocols for their mitigation, with particular emphasis on applications in pharmaceutical analysis and drug development.
Theoretical Background and Definitions
Types of Interferences
In spectrochemical analysis, interferences are broadly classified into two categories: spectral interferences and matrix effects. Spectral interference occurs when the analytical signal of the target analyte is overlapped by a signal from another element or compound present in the sample. In contrast, matrix effects refer to changes in the analyte signal caused by the overall sample composition affecting atomization, excitation, or absorption processes [25].
The distinction between these interference types is visually represented in their effect on calibration curves. Line overlap produces parallel shifts of the calibration curve, always resulting in measured intensities that are higher than the true value. Matrix effects, however, result in a change in the slope of the calibration curve, which can either increase or decrease the measured intensity depending on whether enhancement or absorption dominates [25].
Mathematical Correction Models
Mathematical models for interference correction form the foundation of modern spectrochemical analysis. The basic calibration function follows the form Ci = A0 + A1Ii, where Ci is the concentration of element i and Ii is the measured intensity [25]. The table below summarizes the primary correction approaches for both interference types.
Table 1: Mathematical Correction Models for Spectral Interferences
| Interference Type | Correction Equation | Parameters | Application Examples |
|---|---|---|---|
| Single Element Line Overlap | Ci = A0 + A1 (Ii - hCj) | h = correction factor; Cj = concentration of interfering element | Carbon line at C I 193.07 nm overlapped by aluminum line at Al II 193.1 nm in steel analysis [25] |
| Multiple Element Line Overlap | Ci = A0 + A1 (Ii - ΣhijCj) | hij = correction factor for each interfering element | Z and Z-1 interference in XRF: Kβ of chromium overlaps Kα of manganese [25] |
| Intensity-Based Line Overlap | Ci = A0 + A1 (Ii - ΣhijIj) | Ij = measured intensity of interfering element | Useful when concentration of interfering element is unknown [25] |
| Matrix Effect (Influence Coefficient) | Ci = A0 + A1Ii (1 ± kCj) | k = correction factor; ± indicates enhancement/absorption | Iron absorbs copper X-rays but enhances chromium X-rays in soil analysis [25] |
| Multiple Element Matrix Effect | Ci = A0 + A1Ii (1 ± ΣkijCj) | kij = correction factor for each matrix element | Chromium in steel changes calibration slope for carbon due to carbide formation [25] |
Experimental Protocols for Identification and Correction
Protocol 1: Identification of Interference Type
Purpose: To determine whether a deviation from the base calibration curve is caused by spectral interference or matrix effects.
Materials and Equipment:
- High-resolution spectrophotometer
- Certified reference materials without interferents
- Samples with suspected interferents
- Data processing software
Procedure:
- Establish a base calibration curve using certified standards without interfering elements.
- Analyze samples containing suspected interfering elements.
- Plot results relative to the base curve:
- If data points show parallel shifts to the right (higher intensity), spectral interference is indicated.
- If data points show changed slope (either increased or decreased), matrix effects are indicated.
- Examine spectra visually for obvious line overlaps when possible.
- For matrix effects in XRF, points to the left of the curve indicate absorption, while points to the right indicate enhancement [25].
Troubleshooting:
- If interference type is ambiguous, use standard addition method to confirm.
- For complex matrices, consider using a higher-resolution instrument to separate overlapping signals.
Protocol 2: Matrix Effect Correction in Energy Dispersive X-Ray Fluorescence (EDXRF) for Rock Samples
Purpose: To correct for matrix effects in heterogeneous rock samples using a classification-based approach.
Materials and Equipment:
- Portable EDXRF spectrometer with Ag target (35 kV, 2 μA)
- Rock samples representing different matrix types
- Monte Carlo simulation software (Geant4 toolkit)
- Reference materials for validation [26]
Procedure:
- Sample Preparation:
- Collect representative rock masses (e.g., griotte, syenite, granite, mica schist, andesite, gabbro, limestone, calcite carbonate).
- Ensure homogeneous particle size where possible.
- Prepare thin sections for homogeneous samples.
Monte Carlo Simulation:
- Configure simulation parameters to match portable EDXRF spectrometer with Ag target.
- Define mineralogical compositions for different rock types.
- Simulate X-ray interactions for major elements (Si, Ca, K, Al, Fe, Mg, Na, Ti, Mn, P).
- Calculate matrix effect correction coefficients based on simulation results [26].
Spectrum Measurement:
- Measure EDXRF spectra of rock samples using identical instrument parameters.
- Collect spectra for both major and trace elements.
- Record net peak areas for quantitative analysis.
Matrix Effect Classification:
- Classify rock samples based on matrix effect similarity rather than petrographic classification.
- Apply influence coefficients method for inter-element corrections.
- Use the fundamental parameter method combined with empirical coefficients for final quantification [26].
Validation:
- Compare EDXRF results with reference methods (WD-XRF, ICP-MS, ICP-OES).
- Verify that the same quantification parameters work for different rock types.
Applications and Limitations:
- This method is particularly effective for mineralized rock samples with target element contents of 103-105 mg/kg.
- For samples with low target element contents, applicability must be systematically validated [26].
Advanced Correction Techniques
Background Correction in Atomic Absorption Spectroscopy
Continuum Source Method: This approach uses a deuterium (D2) lamp as a continuum source in addition to the primary hollow cathode lamp. The background absorption is measured with the D2 lamp, while total absorption (analyte + background) is measured with the hollow cathode lamp. The corrected absorbance is obtained by subtracting the D2 lamp absorbance from the hollow cathode lamp absorbance. This method assumes the background absorbance is constant over the wavelength range passed by the monochromator [27].
Zeeman Effect Background Correction: This sophisticated technique applies a magnetic field to the atomizer, which splits the atomic absorption lines into multiple components. A rotating polarizer alternates between measuring absorption at the analytical wavelength (analyte + background) and at a slightly shifted wavelength (background only). The difference provides the corrected analyte absorption. This method is particularly effective for complex matrices and can correct for structured background [27].
Miniaturized Liquid Chromatography as a Solution to Matrix Effects
The transition from conventional HPLC to miniaturized LC (capillary and nanoLC) presents significant advantages for overcoming matrix effects in complex samples. While primarily used in omics sciences, miniaturized LC offers enhanced chromatographic performance and detectability with considerable environmental and economic benefits. The technical barriers to adoption include the need to handle minute sample volumes and lower flow rates, requiring specialized training beyond conventional HPLC expertise [28].
Table 2: Comparison of Interference Correction Methods
| Method | Principle | Advantages | Limitations | Typical Applications |
|---|---|---|---|---|
| Empirical Coefficients | Experimentally derived correction factors | Simple implementation; effective for predictable matrices | Requires many standards; limited to characterized interferences | Routine analysis of similar sample types [26] |
| Fundamental Parameters | Theoretical calculation based on physics of X-ray interactions | Less dependent on standards; applicable to unknown samples | Requires powerful data processing; complex implementation | EDXRF analysis of diverse geological samples [26] |
| Influence Coefficient Methods | Mathematical correction using known inter-element relationships | Comprehensive correction for multiple interferents | Dependent on accurate concentration data | XRF analysis of alloys, soils [25] |
| Monte Carlo Simulation | Computer simulation of photon interactions | Can model complex scenarios without physical samples; accounts for all physical phenomena | Computationally intensive; requires accurate input parameters | Preliminary analysis before experimental verification [26] |
| External Standard Method | Comparison to standards with matched matrix | Direct compensation for matrix effects | Requires appropriate standards; time-consuming | Biological sample analysis [19] |
UFLC-MS/MS: Overcoming Limitations of Spectrophotometry
Donepezil Bioequivalence Study Protocol
Purpose: To demonstrate the advantage of UFLC-MS/MS in overcoming spectral interferences and matrix effects for pharmaceutical analysis.
Materials and Equipment:
- ExionLC AC system with Triple Quad 6500+ mass spectrometer
- Chromolith high resolution RP-18e monolithic column (50 × 4.6 mm)
- Donepezil and Donepezil-d5 (IS) reference standards
- Human plasma samples
- Mobile phases: A (0.1% formic acid in water), B (0.1% formic acid in acetonitrile) [19]
Chromatographic Conditions:
- Column Temperature: Room temperature
- Flow Rate: Multi-stage (3 mL/min initially, reduced to 1.2 mL/min)
- Gradient Program:
- 0-0.1 min: 25% B
- 0.1-0.6 min: 25-60% B
- 0.6-1.1 min: 60-80% B
- 1.1-1.2 min: 80% B
- 1.2-1.5 min: 25% B (re-equilibration)
- Injection Volume: 5 μL
- Total Run Time: 1.5 minutes [19]
Sample Preparation:
- Thaw plasma samples at room temperature.
- Aliquot 200 μL of plasma into 96-well polypropylene plate.
- Add 50 μL of internal standard solution (Donepezil-d5).
- Precipitate proteins with 500 μL methanol.
- Vortex mix and centrifuge at 3,500 g for 5 minutes at 10°C.
- Transfer 200 μL supernatant to another 96-well plate.
- Add 400 μL water and mix.
- Inject 5 μL for LC-MS/MS analysis [19].
Method Validation:
- Linearity: 0.2-50 ng/mL (r > 0.995)
- Intra-day accuracy and precision: Within 15%
- LLOQ: 0.2 ng/mL (S/N > 5)
- Specificity: No interference from plasma components [19]
Comparative Analysis: UFLC-MS/MS vs. Spectrophotometry
The donepezil case study demonstrates key advantages of UFLC-MS/MS over conventional spectrophotometric methods:
Separation Efficiency: The monolithic column provides high-resolution separation of donepezil from potential interferents in just 1.5 minutes, eliminating the need for mathematical corrections of spectral overlaps.
Matrix Effect Handling: Simple protein precipitation effectively removes matrix components that would cause significant interference in atomic spectroscopic methods, eliminating the need for complex matrix-matched standards or mathematical corrections.
Speed and Throughput: The 1.5-minute analysis time represents a significant improvement over conventional HPLC methods (typically ≥4 minutes) and spectrophotometric methods requiring extensive sample preparation and correction procedures [19].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Interference Mitigation
| Reagent/Equipment | Function | Application Example | Considerations |
|---|---|---|---|
| Certified Reference Materials | Establishing base calibration curves; method validation | Quality control in spectrochemical analysis | Must match sample matrix as closely as possible [25] |
| Deuterated Internal Standards | Compensation for matrix effects in MS | Donepezil-d5 for LC-MS/MS bioanalysis | Should elute similarly to analyte; not present in original sample [19] |
| Monolithic LC Columns | High-efficiency separation under high flow rates | Ultrafast separation of donepezil from plasma matrix | Enables faster flow rates without backpressure issues [19] |
| Influence Coefficient Standards | Quantifying inter-element effects | Matrix effect correction in XRF | Must include all significant interelements in expected samples [25] |
| Protein Precipitation Solvents | Removing proteinaceous matrix components | Plasma sample preparation for LC-MS/MS | Organic solvents (methanol, acetonitrile) commonly used [19] |
| Matrix-Matched Standards | Compensating for matrix effects by mimicking sample composition | External standard method in XRF | Preparation requires thorough characterization of sample matrix [26] |
Workflow and Relationship Diagrams
Spectral interference and matrix effects present significant challenges in spectrochemical analysis, requiring sophisticated correction approaches ranging from mathematical models to instrumental solutions. The comparison between traditional spectrophotometric methods and emerging UFLC-MS/MS techniques reveals a paradigm shift in interference management: rather than mathematically correcting for interferences after measurement, chromatographic approaches physically separate analytes from interferents before detection. This fundamental difference underscores the advantage of UFLC-MS/MS for complex pharmaceutical applications where accuracy, sensitivity, and throughput are paramount. As analytical science continues to evolve, the integration of computational methods like Monte Carlo simulation with experimental techniques provides a powerful framework for addressing these perennial analytical challenges, enabling researchers to discriminate between true analyte signals and analytical artifacts with increasing confidence and precision.
The Role of Mass Spectrometry (MS) Detection in Modern LC
The integration of Mass Spectrometry (MS) detection with Liquid Chromatography (LC) has fundamentally transformed analytical capabilities in modern laboratories, particularly within pharmaceutical research and development. This combination provides a powerful tool for the separation, identification, and quantification of compounds in complex mixtures. Within the context of discriminating between ultra-fast liquid chromatography and traditional spectrophotometric methods, LC/MS offers unparalleled specificity and sensitivity. Unlike spectrophotometric detectors which rely on UV-Vis absorbance, MS detection provides direct molecular characterization by measuring the mass-to-charge ratio of analytes, effectively eliminating ambiguity in compound identification [29]. This document details the application of modern LC/MS platforms and provides standardized protocols for their use in drug development.
Current LC/MS Platforms and Technical Specifications
Recent advancements in instrumentation have yielded a new generation of LC and MS systems designed for higher performance, throughput, and ease of use. The table below summarizes key new products introduced between 2024-2025, highlighting their application-specific designs and performance metrics [30].
Table 1: New HPLC/UHPLC and MS Systems (2024-2025)
| Vendor | System/Model | Type | Key Features and Specifications |
|---|---|---|---|
| Agilent | Infinity III LC Series | UHPLC | Pressures up to 1300 bar; Bio-inert flow paths for extreme pH stability; Modules for automated method development and impurity analysis [30]. |
| Shimadzu | i-Series | HPLC/UHPLC | Compact, integrated design; Pressure capability up to 70 MPa (10,152 psi); Eco-friendly reduced energy consumption; Supports a wide range of detectors [30]. |
| Thermo Fisher Scientific | Vanquish Neo UHPLC | UHPLC | Tandem direct injection workflow with two-pump, two-column configuration for parallel analysis, increasing throughput and reducing carryover [30]. |
| Waters | Alliance iS Bio HPLC | HPLC | Tailored for biopharma QC; Pressures up to 12,000 psi; MaxPeak HPS technology; Bio-inert design for a pH range of 1-13 [30]. |
| Sciex | 7500+ MS/MS | Triple Quadrupole | Enhanced resilience and user serviceability; 900 MRM/sec capability; Mass Guard technology and DJet+ interface; Compatible with energy-saving dry pumps [30]. |
| Bruker | timsTOF Ultra 2 | Trapped Ion Mobility-TOF | Advanced 4D proteomics for deep, high-fidelity analysis; Capable of measuring >1000 proteins from a 25-pg sample [30]. |
| Sciex | ZenoTOF 7600+ | High-Resolution MS | Zeno Trap Technology and Electron Activated Dissociation (EAD); High-speed scanning up to 640 Hz for advanced proteomics and biomarker research [30]. |
Comparative Quantitative Mass Spectrometry Platforms
The selection of an appropriate MS acquisition method is critical for the success of any experiment, especially when dealing with low-abundance analytes in complex matrices. A comparative study of four primary quantitative MS platforms illustrates their respective strengths and optimal use cases [31].
Table 2: Comparison of Quantitative MS Acquisition Platforms
| Acquisition Platform | Key Principle | Strengths | Considerations | Ideal Use Case |
|---|---|---|---|---|
| LC-MRM (Multiple Reaction Monitoring) | Targeted analysis using a triple-quadrupole MS to monitor specific precursor/fragment ion pairs [31]. | High sensitivity and specificity; Excellent quantitative precision; Considered the gold standard for targeted quantification [31]. | Requires a priori knowledge of targets to develop methods; Lower resolution can lead to interference. | Validated, high-throughput quantitative assays for known compounds (e.g., pharmacokinetics) [31]. |
| LC-PRM (Parallel Reaction Monitoring) | Targeted precursor selection with high-resolution, accurate-mass MS/MS detection [31]. | High specificity from accurate mass fragments; Reduced interference compared to MRM; No need for pre-defined fragment ions [31]. | Similar to MRM, requires a predefined list of target precursors. | Targeted quantification where high resolution is needed to eliminate background interference [31]. |
| LC-MS/MS with DDA (Data-Dependent Acquisition) | "Discovery" mode; full scan survey with automatic selection of abundant ions for MS/MS [31]. | Ideal for untargeted discovery and protein/peptide identification. | Susceptible to undersampling of low-abundance ions; Can result in significant missing data across samples. | Preliminary discovery phases to identify components in unknown mixtures [31]. |
| LC-MS/MS with DIA (Data-Independent Acquisition) | Sequential fragmentation of all ions in pre-defined, wide m/z windows [31]. | More consistent and comprehensive peptide detection than DDA; Reduced missing data; Allows retrospective data mining [31]. | Generates complex, chimeric spectra that require specialized software and spectral libraries for deconvolution. | Large-scale quantitative proteomic studies where comprehensive data capture is essential [31]. |
Application Note: Superiority of LC-MS in Toxicological Screening
Background and Objective
Traditional immunoassay (IA)-based drug screens are limited by antibody cross-reactivity, restricting the scope of analysis and leading to potential false positives or negatives. This application note summarizes a 2025 study comparing a comprehensive LC-MS screen with conventional IA techniques in whole blood [32].
Experimental Protocol
- Samples: 919 adjudicated whole blood specimens previously analyzed by ELISA and EMIT immunoassays [32].
- Sample Preparation: Supported Liquid Extraction (SLE) was used for clean-up and analyte concentration [32].
- Instrumentation: Analysis was performed using Liquid Chromatography-Quadrupole Time-of-Flight-Mass Spectrometry (LC-QTOF-MS) [32].
- Data Analysis: The high-resolution MS data was interrogated retrospectively.
Results and Discussion
The HRMS-based screen identified an additional 709 positive drug findings, encompassing 67 different compounds that were not detected by the initial immunoassays [32]. This highlights a significant limitation of IA-based methods, particularly with the emergence of new therapeutics and new psychoactive substances (NPS) for which specific antibodies may not exist. The study underscores the key advantages of LC-HRMS:
- Enhanced Specificity: Direct identification based on mass, avoiding antibody cross-reactivity.
- Broader Scope: Ability to screen for a vast number of compounds in a single method.
- Retrospective Analysis: Stored data can be re-interrogated for new compounds without re-running samples [32].
Detailed Protocol: Determination of a Nitrosamine Impurity by UHPLC-MS/MS
Background
This protocol details a validated method for determining the genotoxic impurity N-Nitrosoduloxetine in Duloxetine HCl active pharmaceutical ingredient (API) using UHPLC-MS/MS, demonstrating the application of LC-MS for sensitive and specific impurity testing [33].
Materials and Reagents
Table 3: Research Reagent Solutions and Essential Materials
| Item | Function / Specification |
|---|---|
| Duloxetine HCl API | The drug substance to be tested for the impurity [33]. |
| N-Nitrosoduloxetine Reference Standard | Used for method qualification, calibration, and quantification [33]. |
| Acetonitrile (MS Grade) | Organic mobile phase component [33]. |
| Formic Acid 0.1% in Water | Aqueous mobile phase component; aids in ionization [33]. |
| Waters Acquity HSS T3 Column | (3.0 × 100 mm, 1.8 μm) for chromatographic separation [33]. |
Instrumentation and Method Parameters
- System: Ultra-High-Performance Liquid Chromatography system coupled to a tandem mass spectrometer with electrospray ionization (ESI) [33].
- Chromatography:
- Column: Acquity HSS T3 (3.0 × 100 mm, 1.8 μm).
- Mobile Phase: Formic acid 0.1% in water combined with acetonitrile (gradient elution).
- Run Time: 11 minutes [33].
- Mass Spectrometry:
- Ionization: Electrospray Ionization (ESI).
- Mode: Multiple Reaction Monitoring (MRM) for high sensitivity and selectivity.
Sample Preparation
- Both reference and sample solutions are prepared using a simple dissolution protocol [33].
Validation Data
The method was validated as per regulatory guidelines (e.g., ICH) demonstrating [33]:
- Limit of Detection (LOD): 0.7 ppb (parts-per-billion).
- Limit of Quantification (LOQ): 70 ppb.
- Trueness (Recovery): 100% - 110%.
- Linearity: Regression coefficients (R) of 0.9990 - 0.9991.
Visualized Workflows
LC-MS Analysis and Data Processing
Targeted vs. Discovery MS Strategies
Practical Applications: Implementing UFLC and Spectrophotometry in the Lab
In the pharmaceutical industry, the selection of an analytical technique for drug assay is a critical decision that impacts the efficiency, cost, and environmental footprint of quality control operations. This research focuses on the methodological discrimination between ultra-fast liquid chromatography (UFLC) and modern spectrophotometric techniques for the quantitative analysis of active pharmaceutical ingredients (APIs) in both bulk and formulated products. The evolution of analytical science has led to significant advancements in both instrumental categories, with UFLC offering exceptional separation power and sensitivity, while contemporary spectrophotometric methods employ sophisticated mathematical processing to resolve complex mixtures without physical separation [34] [30].
The core of this technical evaluation centers on identifying the appropriate application domains for these techniques based on analytical requirements, matrix complexity, and operational constraints. While chromatographic methods, particularly those coupled with mass spectrometry, provide unparalleled specificity for complex matrices, recent spectrophotometric approaches have demonstrated remarkable capability in analyzing multi-component formulations through mathematical resolution of overlapping spectra [35] [36]. This application note provides a structured comparison, detailed experimental protocols, and practical guidance to inform method selection in pharmaceutical research and development.
Modern Spectrophotometric Methods
Ultraviolet-Visible (UV-Vis) spectrophotometry remains a cornerstone of pharmaceutical analysis due to its simplicity, cost-effectiveness, and minimal solvent consumption. Modern implementations have evolved beyond simple absorbance measurement to incorporate sophisticated mathematical processing that enables simultaneous determination of multiple analytes despite significant spectral overlap [35] [37].
Key Advanced Spectrophotometric Techniques:
- Successive Ratio Subtraction with Constant Multiplication (SRS-CM): Enables component quantification at respective absorption maxima through sequential spectral manipulation [35]
- Successive Derivative Subtraction with Constant Multiplication (SDS-CM): Utilizes first-derivative spectra for enhanced resolution of overlapping peaks [35]
- Ratio Difference Spectrophotometry: Measures amplitude differences in ratio spectra at strategically selected wavelengths [37]
- Dual-Wavelength Resolution: Calculates component concentrations using absorptivity factors at multiple wavelengths [37]
These mathematical spectrophotometric methods effectively resolve binary and ternary mixtures without requiring physical separation, making them particularly valuable for routine quality control of fixed-dose combination products [38] [36].
Ultra-Fast Liquid Chromatography (UFLC) and LC-MS
Ultra-fast liquid chromatography represents the cutting edge of separation science, leveraging sub-2μm particle columns operating at high pressures (up to 1300 bar) to achieve rapid separations with superior resolution [34] [30]. When coupled with mass spectrometry (LC-MS), the technique provides exceptional specificity and sensitivity for complex pharmaceutical analyses.
Recent Technological Advancements:
- Monolithic columns enable high flow rates (3 mL/min) with minimal backpressure, reducing analysis times to under 1.5 minutes for some applications [19]
- Multi-stage flow rate programming optimizes separation efficiency while conserving mobile phase [19]
- Advanced detection systems including tandem mass spectrometry with multiple reaction monitoring (MRM) provide unparalleled specificity [19]
- Biocompatible systems with MaxPeak HPS technology facilitate analysis of biomolecules without adsorption issues [30]
The integration of intelligent chromatography data systems (CDS) with remote operation capabilities further enhances method reproducibility and operational efficiency [30].
Comparative Performance Data
Table 1: Analytical Performance Characteristics of Spectrophotometric vs. Chromatographic Methods
| Parameter | Spectrophotometric Methods | UFLC Methods | LC-MS/MS Methods |
|---|---|---|---|
| Linear Range | 1-100 μg/mL [35] [36] | 5-50 μg/mL [39] | 0.2-50 ng/mL [19] |
| Analysis Time | 1-5 minutes [38] [37] | 1.5-4 minutes [19] [39] | 1.5-4 minutes [19] |
| Limit of Detection | 0.26-0.92 μg/mL [36] | ~1 μg/mL [39] | 0.2 ng/mL [19] |
| Precision (%RSD) | <1.5% [36] [37] | <1.5% [39] | <15% [19] |
| Accuracy (%Recovery) | 98-102% [35] [37] | 99.7-100.3% [39] | 85-115% [19] |
| Solvent Consumption | 10-50 mL/day [35] | 500-1000 mL/day | 500-1000 mL/day |
| Instrument Cost | Low | High | Very High |
Table 2: Application Domains for Pharmaceutical Analysis
| Analysis Type | Recommended Technique | Justification |
|---|---|---|
| Routine QC of Solid Dosage Forms | Spectrophotometric methods [35] [39] | Cost-effective, rapid, sufficient accuracy |
| Bioequivalence Studies | LC-MS/MS [19] | Required sensitivity for plasma samples |
| Fixed-Dose Combination Products | Mathematical spectrophotometry [35] [36] | Resolves overlapping spectra without separation |
| Stability-Indicating Methods | HPLC/UFLC [39] | Separates degradants from API |
| Trace Analysis | LC-MS/MS [34] [19] | Superior sensitivity and specificity |
| Green Analytical Chemistry | Spectrophotometry [35] [36] | Minimal organic solvent consumption |
Experimental Protocols
Protocol 1: Simultaneous Assay of Ternary Mixture by Mathematical Spectrophotometry
This protocol details the simultaneous determination of Telmisartan (TEL), Chlorthalidone (CHT), and Amlodipine (AML) using Successive Ratio Subtraction coupled with Constant Multiplication (SRS-CM) [35].
Materials and Reagents:
- Reference standards: TEL, CHT, AML (purity >98%)
- Solvent: Ethanol (HPLC grade)
- Instrument: Double-beam UV-Vis spectrophotometer with 1 cm quartz cells
Procedure:
- Stock Solution Preparation: Accurately weigh 50 mg of each standard and transfer to separate 100 mL volumetric flasks. Dissolve and dilute to volume with ethanol to obtain 500 μg/mL stock solutions.
- Working Standard Preparation: Pipette 20 mL from each stock solution into separate 100 mL volumetric flasks. Dilute to volume with ethanol to obtain 100 μg/mL working standards.
- Calibration Curve Construction:
- Prepare serial dilutions covering 5-40 μg/mL for TEL, 10-100 μg/mL for CHT, and 5-25 μg/mL for AML.
- Scan zero-order absorption spectra from 200-400 nm against ethanol blank.
- Record absorbances at 295.7 nm (TEL), 275.0 nm (CHT), and 359.5 nm (AML).
- Plot absorbance vs. concentration for each drug and compute regression equations.
- Sample Preparation:
- Finely powder 20 tablets and accurately weigh powder equivalent to one tablet.
- Transfer to 100 mL volumetric flask, add 70 mL ethanol, sonicate for 15 minutes.
- Dilute to volume with ethanol and filter.
- Dilute filtrate appropriately to fall within calibration range.
- Calculation:
- Measure absorbances at the three wavelengths.
- Compute concentrations using the respective regression equations.
Method Validation:
- Establish linearity (r² > 0.999), precision (%RSD < 2%), and accuracy (98-102% recovery)
- Determine LOD and LOQ using signal-to-noise ratio approach
Protocol 2: Ultra-Fast LC-MS/MS Analysis for Bioequivalence Studies
This protocol describes the determination of donepezil in human plasma using UFLC-MS/MS with monolithic column technology [19].
Materials and Reagents:
- Reference standards: Donepezil and Donepezil-d5 (IS)
- Solvents: HPLC-grade methanol, acetonitrile, formic acid
- Biological matrix: K2EDTA human plasma
- Equipment: UFLC system coupled to triple quadrupole mass spectrometer
Chromatographic Conditions:
- Column: Chromolith high resolution RP-18e monolithic column (50 × 4.6 mm)
- Mobile Phase: A: 0.1% formic acid in water; B: 0.1% formic acid in acetonitrile
- Gradient Program: 25% B (0.1 min) → 60% B (0.6 min) → 80% B (1.1 min)
- Flow Rate: Multi-stage: 3 mL/min (0.1 min) → 1.2 mL/min (0.6-1.1 min) → 3 mL/min (equilibration)
- Injection Volume: 5 μL
- Run Time: 1.5 minutes
Mass Spectrometric Conditions:
- Ionization: ESI positive mode
- MRM Transitions: m/z 380 → 91 (donepezil), m/z 385 → 96 (IS)
- Ion Spray Voltage: 5500 V
- Source Temperature: 600°C
- Collision Energy: 40 eV
Sample Preparation:
- Thaw frozen plasma samples at room temperature
- Aliquot 200 μL plasma into 96-well polypropylene plate
- Add 50 μL IS working solution (donepezil-d5 in 50% methanol)
- Precipitate proteins with 500 μL methanol
- Vortex mix for 3 minutes, then centrifuge at 3500 × g for 5 minutes at 10°C
- Transfer 200 μL supernatant to new 96-well plate, add 400 μL water
- Inject 5 μL into LC-MS/MS system
Method Validation:
- Demonstrate specificity using six different lots of blank plasma
- Establish calibration curve over 0.2-50 ng/mL with r² > 0.995
- Evaluate intra-day and inter-day precision (%RSD < 15%) and accuracy (85-115%)
- Assess matrix effect, recovery, and stability under various conditions
Visualized Workflows
Diagram 1: Analytical Method Selection and Workflow Comparison. The decision pathway illustrates technique selection based on sample complexity and analytical requirements.
Research Reagent Solutions
Table 3: Essential Materials and Reagents for Pharmaceutical Analysis
| Item | Specification | Application | Key Considerations |
|---|---|---|---|
| HPLC-Grade Solvents | Methanol, acetonitrile, ethanol | Mobile phase preparation | Low UV cutoff, minimal impurities [35] [19] |
| Chromatographic Columns | C18 (sub-2μm, monolithic) | UFLC separations | High pressure stability, reproducibility [19] [30] |
| Reference Standards | Certified purity (>98%) | Calibration, quantification | Traceable to reference materials [35] [19] |
| Volumetric Glassware | Class A precision | Solution preparation | Certified accuracy, minimal uncertainty [35] [39] |
| Sample Filtration | 0.22-0.45 μm membranes | Particulate removal | Compatibility with analytes and solvents [39] |
| Mass Spectrometry Additives | Formic acid, ammonium salts | LC-MS mobile phase modifiers | High purity, minimal background [19] |
The comparative analysis presented in this application note demonstrates that both spectrophotometric and chromatographic techniques occupy distinct and valuable positions within the pharmaceutical analytical workflow. The selection between these methodologies should be guided by specific analytical requirements, matrix complexity, and operational constraints.
Key Discrimination Factors:
Analysis Complexity: For simple formulations with minimal excipient interference, mathematical spectrophotometric methods provide excellent results with significantly reduced operational costs and analysis time [35] [37]. The implementation of techniques such as derivative spectroscopy and ratio subtraction enables effective resolution of multi-component systems without physical separation.
Sensitivity Requirements: UFLC-MS/MS delivers superior sensitivity (ng/mL to pg/mL range) essential for bioequivalence studies, metabolite profiling, and trace analysis [34] [19]. The enhanced separation power of monolithic columns coupled with selective detection addresses the challenges of complex biological matrices.
Green Chemistry Considerations: Recent emphasis on sustainable analytical practices has highlighted the ecological advantages of spectrophotometric methods, which typically consume minimal organic solvents and generate less hazardous waste [35] [36]. Assessment tools such as AGREE, GAPI, and BAGI provide quantitative metrics for environmental impact evaluation.
Throughput and Operational Efficiency: While UFLC provides exceptional analytical power, spectrophotometry offers superior throughput for routine quality control applications, with analysis times frequently under five minutes and minimal sample preparation [38] [39].
In conclusion, the discrimination between ultra-fast liquid chromatography and spectrophotometric methods for drug assay requires careful consideration of the specific analytical challenge. Spectrophotometric approaches present an optimal solution for routine quality control of pharmaceutical formulations, particularly fixed-dose combinations, while UFLC and LC-MS/MS remain indispensable for complex matrices requiring high sensitivity and specificity. The continued advancement in both instrumental categories ensures that researchers have access to increasingly sophisticated tools for comprehensive pharmaceutical analysis.
High-Throughput Analysis and Metabolite Identification with UFLC-MS
Ultra-Fast Liquid Chromatography coupled with Mass Spectrometry (UFLC-MS) has emerged as a cornerstone technology in modern analytical science, particularly for high-throughput metabolomic studies and comprehensive metabolite profiling. This advanced instrumentation addresses the critical need for rapid, sensitive, and selective analysis of complex biological samples, enabling researchers to decipher metabolic pathways and identify biomarkers with unprecedented efficiency. The transition from conventional spectrophotometric methods to sophisticated UFLC-MS platforms represents a paradigm shift in analytical capabilities, offering superior resolution, faster analysis times, and enhanced sensitivity for characterizing diverse metabolite classes. Within pharmaceutical development and clinical diagnostics, UFLC-MS provides the necessary analytical power to unravel complex biochemical interactions, monitor therapeutic interventions, and advance personalized medicine initiatives through comprehensive metabolite identification and quantification.
Performance Comparison: UFLC-MS vs. Traditional Methods
The analytical superiority of UFLC-MS systems becomes evident when comparing key performance metrics against traditional spectrophotometric and conventional chromatographic methods. The following tables summarize quantitative performance data from validated UFLC-MS methods across various applications.
Table 1: Analytical Performance Metrics of UFLC-MS Methods in Metabolite Analysis
| Performance Parameter | Traditional Spectrophotometry | Conventional HPLC | UFLC-MS/MS |
|---|---|---|---|
| Analysis Time | ~2 hours (IEC with ninhydrin) [40] | 30-60 minutes | 7.5-19 minutes [40] [41] |
| Sensitivity (LOQ) | Micromolar range | Nanomolar range | 0.05-5 ng/mL (drugs) [41], ≤2.5 μmol/L (amino acids) [40] |
| Throughput | Low (limited sample capacity) | Moderate | High (19-min analysis) [40] |
| Multi-analyte Capacity | Limited (often single analyte) | Moderate | High (115+ simultaneously) [41] |
| Structural Information | None | Limited | Comprehensive (fragmentation patterns) |
Table 2: Validation Parameters for UFLC-MS Metabolite Identification Methods
| Validation Parameter | Reported Performance | Application Context |
|---|---|---|
| Precision (CV) | <10% inter-assay [40], <5% RSD [42] | Amino acid quantification [40] |
| Accuracy (Recovery) | 77-160% [42] | Pharmaceutical contaminants in water [42] |
| Linearity (R²) | ≥0.999 [42] | Pharmaceutical contaminants [42] |
| Correlation (CCC) | >0.99 for 10/23 amino acids [40] | Method comparison to IEC [40] |
| Calibration Stability | <5% bias over 12 weeks [40] | Clinical amino acid analysis [40] |
The data demonstrate that UFLC-MS methods achieve significantly reduced analysis times while maintaining excellent precision and accuracy. For example, a validated UFLC-MS method for amino acid analysis completed separations in 19 minutes compared to approximately 2 hours for traditional ion-exchange chromatography with post-column ninhydrin derivatization [40]. This 6-fold improvement in throughput enables rapid processing of large sample batches essential for clinical and pharmaceutical applications.
Experimental Protocols
Sample Preparation for Global Metabolomics
Principles: Appropriate sample preparation is critical for comprehensive metabolite extraction while minimizing matrix effects. The protocol must preserve metabolic integrity and ensure compatibility with UFLC-MS analysis.
Protocol Steps:
- Protein Precipitation: Add 300-400 μL of cold methanol:acetonitrile (1:1, v/v) or 2:1 (v/v) to 100 μL of biological sample (plasma, serum, or cell culture supernatant) [41] [43].
- Vortexing and Centrifugation: Vortex mix for 30-60 seconds, then centrifuge at 14,000-16,000 × g for 15 minutes at 4°C to pellet proteins.
- Supernatant Collection: Transfer clarified supernatant to a new vial for direct injection or evaporate to dryness under nitrogen stream.
- Reconstitution: Reconstitute dried extracts in 100-200 μL of initial mobile phase compatible with UFLC separation (typically water:acetonitrile or methanol with 0.1% formic acid) [43] [44].
- Centrifugation and Transfer: Centrifuge again at 14,000 × g for 10 minutes and transfer to MS-compatible vials with inserts.
Special Considerations: For "dilute-and-shoot" approaches, minimal preparation may involve simply diluting urine samples 1:3 with organic solvent mixture (methanol:acetonitrile, 3:1, v/v) followed by centrifugation and direct injection [41].
UFLC-MS Instrumental Parameters
Principles: Optimal chromatographic separation and detection conditions are essential for resolving complex metabolite mixtures and achieving confident identifications.
Protocol Steps:
- Chromatographic System:
- Column: C18 reverse-phase column (100 × 2.1 mm, 1.7-1.8 μm particle size) for small molecule separation [42] [44].
- Mobile Phase: A: Water with 0.1% formic acid; B: Acetonitrile or methanol with 0.1% formic acid.
- Gradient Program: 5-95% B over 7.5-19 minutes depending on application [40] [41].
- Flow Rate: 0.3-0.4 mL/min [44].
- Column Temperature: 40-45°C.
- Injection Volume: 2-10 μL.
- Mass Spectrometric Detection:
- Ionization Source: Electrospray ionization (ESI) in positive or negative mode, depending on target metabolites.
- Source Parameters: Capillary voltage: 3.0 kV; Source temperature: 150°C; Desolvation temperature: 500°C; Cone gas flow: 50 L/hr; Desolvation gas flow: 800 L/hr [44].
- Acquisition Mode: Full scan (m/z 50-1000) for untargeted analysis or Multiple Reaction Monitoring (MRM) for targeted quantification.
- Collision Energies: Optimized for specific metabolite classes (typically 10-40 eV).
Metabolite Identification Workflow
Principles: Confident metabolite identification requires a systematic approach combining accurate mass measurement, fragmentation pattern analysis, and database searching.
Protocol Steps:
- Data Preprocessing: Perform peak detection, alignment, and normalization using software such as XCMS, MZmine, or Progenesis QI.
- Accurate Mass Analysis: Calculate potential elemental compositions for features of interest with mass error <5 ppm.
- Fragmentation Pattern Analysis: Interpret MS/MS spectra to identify characteristic fragment ions and neutral losses.
- Database Searching: Query metabolic databases (KEGG, HMDB, METLIN, PubChem) using both exact mass and fragmentation patterns [45].
- Pathway Mapping: Integrate identified metabolites into biochemical pathways using tools such as MetaboAnalyst, IMPaLA, or ProCyc [43] [45].
- Validation: Confirm identities using authentic standards when available, comparing retention times and fragmentation patterns.
Metabolite Identification Strategies
The identification of metabolites from UFLC-MS data employs a multi-tiered approach that leverages both experimental data and bioinformatic resources. Advanced strategies include:
Genome-Restricted Identification: Metabolome Searcher and similar tools enable putative compound identification by restricting possible matches to metabolites that a specific organism can produce based on its genomic capacity [45]. This approach significantly reduces false positives by incorporating biological context into the identification process.
Tandem MS Spectral Libraries: Experimental MS/MS spectra are matched against reference libraries such as MassBank, GNPS, and NIST MS/MS libraries. The presence of characteristic fragment ions provides structural information about functional groups and molecular substructures.
Retention Time Prediction: Quantitative Structure-Retention Relationship (QSRR) models help predict retention times for candidate structures, adding an additional orthogonal parameter for confident identification.
Metabolic Pathway Mapping: Identified metabolites are mapped onto known biochemical pathways using databases such as KEGG and MetaCyc, providing biological context and helping to identify related metabolites that may have been missed in initial analysis [43] [45]. In dioscin-treated rectal cancer cells, for example, metabolomic analysis revealed significant alterations in 22 metabolites and 8 highly correlated pathways including D-glutamine and D-glutamate metabolism, pyruvate metabolism, and the TCA cycle [43].
Diagram Title: UFLC-MS Metabolite Identification Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of UFLC-MS-based metabolomics requires specific reagents and materials optimized for high-performance separation and detection. The following table details essential research reagent solutions for metabolite analysis.
Table 3: Essential Research Reagent Solutions for UFLC-MS Metabolite Analysis
| Reagent/Material | Function/Purpose | Application Example | Performance Considerations |
|---|---|---|---|
| AccQ•Tag Ultra Derivatization Reagent | Amino acid derivatization for improved LC-MS detection sensitivity | Plasma amino acid quantification for inborn error screening [40] | Enables 19-min analysis with CV<10% for most amino acids [40] |
| C18 Reverse-Phase Columns (1.7-1.8 μm) | Small molecule separation with high efficiency under UHPLC conditions | General metabolomics, pharmaceutical analysis [41] [42] | Enables >2000 injections with good retention time stability (<0.4%) [40] |
| Methanol/Acetonitrile (LC-MS Grade) | Protein precipitation, mobile phase component | Sample preparation for global metabolomics [41] [43] | Minimal background interference, high purity for sensitive detection |
| Formic Acid (LC-MS Grade) | Mobile phase additive for improved ionization in positive mode | General metabolomics in ESI+ mode [43] [44] | 0.1% concentration typically optimal for ESI efficiency |
| Solid Phase Extraction Cartridges (C18, HILIC, mixed-mode) | Sample clean-up and metabolite enrichment | Urine drug screening, targeted metabolomics [42] | Reduces matrix effects, improves sensitivity for trace analytes |
| Stable Isotope-Labeled Internal Standards | Normalization of extraction and ionization variability | Quantitative metabolomics, pharmacokinetic studies [44] | Corrects for matrix effects, enables precise quantification |
Diagram Title: Metabolite Identification Strategies
UFLC-MS technology has revolutionized high-throughput metabolite analysis by delivering unparalleled speed, sensitivity, and selectivity compared to traditional spectrophotometric and conventional chromatographic methods. The detailed protocols and analytical strategies presented in this application note provide researchers with a robust framework for implementing UFLC-MS in diverse experimental contexts, from clinical diagnostics to pharmaceutical development. By leveraging optimized sample preparation techniques, sophisticated instrumental parameters, and advanced bioinformatic tools for metabolite identification and pathway mapping, scientists can fully exploit the capabilities of UFLC-MS platforms to advance metabolomic research and accelerate biomarker discovery. As the field continues to evolve, integration of genomic context and artificial intelligence-assisted identification promises to further enhance the power and precision of UFLC-MS-based metabolite analysis.
Dissolution Testing and Stability-Indicating Methods
In pharmaceutical development, dissolution testing and stability-indicating methods are critical analytical procedures that ensure drug product quality, performance, and stability throughout the shelf life. Dissolution testing measures the rate and extent of active pharmaceutical ingredient (API) release from solid dosage forms under standardized conditions, providing crucial insights into in vivo performance potential [46]. Stability-indicating methods are validated analytical procedures that quantitatively measure the API without interference from degradation products, process impurities, excipients, or other potential components [47] [48]. These methods must demonstrate specificity to detect changes in the active ingredient concentration over time, making them essential for stability studies and shelf-life determination [47].
Within modern analytical research, a significant focus has emerged on comparing ultra-fast liquid chromatography (UFLC) with traditional spectrophotometric techniques for these applications. UFLC offers superior separation capabilities, speed, and specificity, while spectrophotometric methods provide cost-effectiveness and operational simplicity [49]. The discrimination between these methodologies forms a critical research axis in pharmaceutical analysis, particularly for optimizing regulatory submission strategies and quality control workflows.
Core Principles and Regulatory Framework
Dissolution Testing Fundamentals
Dissolution testing serves as a vital performance indicator for solid oral dosage forms. The fundamental principle involves placing a dosage form in a dissolution medium under controlled temperature and agitation, with samples withdrawn at specified intervals to quantify the amount of drug dissolved [46]. The test aims to have "sink conditions," where the medium volume is at least three times that required to form a saturated solution of the drug substance, ensuring results accurately reflect dosage form properties rather than solubility limitations [46].
The Biopharmaceutics Classification System (BCS) provides a scientific framework for applying dissolution testing. For BCS Class II drugs like atorvastatin (low solubility, high permeability), dissolution rate is the primary limiting factor for absorption, making in vitro-in vivo correlation (IVIVC) particularly valuable [50]. A well-designed dissolution method must be discriminating, capable of detecting significant changes in formulation composition or manufacturing process that could affect in vivo performance [50] [46].
Stability-Indicating Method Requirements
According to ICH guidelines, stability-indicating methods must be properly validated for sensitivity, specificity, accuracy, reliability, reproducibility, and robustness [47]. The United States Food and Drug Administration defines these methods as validated quantitative analytical procedures that can detect changes in drug substance concentration over time without interference from degradation products, excipients, or other components [47].
These methods are developed through forced degradation studies, where the drug substance is stressed under various conditions including hydrolysis (acid and base), oxidation, thermal, and photolytic exposure [51]. The resulting samples are analyzed to demonstrate method specificity and the ability to separate degradation products from the main API [48].
Figure 1: Development workflow for stability-indicating methods, highlighting forced degradation studies as a critical component [51].
Analytical Methodologies: UFLC versus Spectrophotometry
Ultra-Fast Liquid Chromatography (UFLC/UPLC)
UFLC, also called Ultra Performance Liquid Chromatography (UPLC), represents a significant advancement in liquid chromatography, utilizing sub-2μm particles and higher operating pressures to achieve superior resolution, speed, and sensitivity compared to conventional HPLC [51] [52]. The stability-indicating capability of UFLC makes it particularly valuable for pharmaceutical analysis, as demonstrated in the determination of diclofenac sodium with rapid analysis time (1.2 minutes) while effectively separating degradation products formed under stress conditions [52].
For ticlopidine hydrochloride analysis, a stability-indicating UPLC method was developed using a Zorbax SB-C18 column (50 mm × 4.6 mm, 1.8 μm) with methanol-0.01 M ammonium acetate buffer (pH 5.0) in the ratio 80:20 v/v as mobile phase at a flow rate of 0.8 mL/min [51]. The method demonstrated excellent linearity (62.5–375 μg/mL), precision (RSD <1.31%), and accuracy (recovery 98.80–101.50%), successfully separating degradation products from the main peak [51].
Spectrophotometric Methods
Spectrophotometric techniques offer simpler, more cost-effective alternatives for dissolution testing, though with limited specificity for stability-indicating applications. For the analysis of mirabegron and solifenacin combination therapy, spectrophotometric methods including first derivative ratio spectrophotometry (measuring at 224.8 nm and 258.6 nm, respectively) and extended ratio subtraction method have been successfully applied [49]. These methods effectively resolve spectral overlaps in binary mixtures, providing environmentally friendly alternatives to chromatographic methods [49].
UV spectrophotometry has also been utilized for dissolution testing of atorvastatin tablets, where it provided adequate quantification for the dissolution application, though with potentially less specificity than chromatographic methods for stability assessment [50].
Comparative Analysis
Table 1: Comparison of UFLC and Spectrophotometric Methods for Pharmaceutical Analysis
| Parameter | UFLC/UPLC | Spectrophotometry |
|---|---|---|
| Separation Capability | Excellent - separates API from impurities and degradants [52] | Limited - no separation, measures total absorbance [49] |
| Analysis Time | Fast (1-5 minutes typical) [52] | Very fast (minutes) [49] |
| Specificity | High - through separation and peak identification [51] | Moderate - dependent on spectral differences [49] |
| Sensitivity | Excellent (LOD ~2 ppm demonstrated) [52] | Good for main component, limited for impurities |
| Cost | Higher (equipment, solvents, columns) [49] | Lower (minimal solvent use, simple equipment) [49] |
| Stability-Indicating | Yes - validated for forced degradation studies [51] [52] | Limited - not typically suitable for stability studies |
| Environmental Impact | Higher solvent consumption [49] | Lower - considered green alternative [49] |
| Application in Dissolution | Preferred for complex formulations [46] | Suitable for simple formulations without interference [50] |
Experimental Protocols
Development and Validation of a Discriminative Dissolution Method for Atorvastatin Tablets
Objective: To develop and validate a discriminative dissolution method for atorvastatin tablets capable of establishing IVIVC [50].
Materials and Equipment:
- Dissolution apparatus (USP Apparatus 2, paddle type)
- UPLC/HPLC system with UV detection
- Atorvastatin calcium reference standard
- Test formulations and placebo
- Potassium phosphate buffer (pH 6.0)
- Reverse-phase C18 column (250 × 4.6 mm; 5 μm)
Procedure:
- Solubility and Sink Conditions: Determine solubility in various media including 0.1 M HCl, gastric fluid (pH 1.2), citrate buffer (pH 3.0), acetate buffer (pH 4.0), phosphate buffers (pH 5.0, 6.0, 6.8), and water. Select medium providing sink conditions [50].
- Dissolution Test Parameters:
- Apparatus: USP II (paddle)
- Medium: 900 mL potassium phosphate buffer, pH 6.0
- Temperature: 37 ± 0.5°C
- Rotation speed: 50 rpm
- Sampling times: 5, 10, 15, 20, 30, 45, 60, 120 minutes [50]
- Sample Analysis:
- Withdraw samples through 35-μm filter
- Analyze by UPLC with mobile phase sodium acetate buffer (pH 4.2):acetonitrile (45:55 v/v)
- Flow rate: 1.0 mL/min, detection: 245 nm [50]
- IVIVC Development:
- Obtain in vivo data from bioequivalence study
- Calculate fraction absorbed using Loo-Riegelman method
- Correlate with dissolved fraction using appropriate scale factor [50]
Stability-Indicating UPLC Method for Ticlopidine Hydrochloride
Objective: To develop and validate a stability-indicating UPLC method for ticlopidine hydrochloride in solid dosage forms [51].
Materials and Equipment:
- UPLC system with PDA detector
- Zorbax SB-C18 column (50 mm × 4.6 mm, 1.8 μm)
- Ticlopidine hydrochloride standard and tablet formulation
- Ammonium acetate, glacial acetic acid, methanol (UPLC grade)
- Hydrochloric acid, sodium hydroxide, hydrogen peroxide (reagent grade)
Chromatographic Conditions:
- Mobile phase: Methanol-0.01 M ammonium acetate buffer, pH 5.0 (80:20, v/v)
- Flow rate: 0.8 mL/min
- Injection volume: 4.0 μL
- Detection: 235 nm
- Column temperature: Ambient [51]
Forced Degradation Studies:
- Acid Degradation: Heat drug content in 0.1 N HCl at 80°C for 2 hours, then neutralize [51].
- Alkali Degradation: Heat drug content in 1 M NaOH at 80°C for 2 hours, then neutralize [51].
- Oxidative Degradation: Heat drug content in 3% H₂O₂ at 80°C for 1 hour [51].
- Thermal Degradation: Expose solid drug at 80°C for 72 hours [51].
- Photolytic Degradation: Expose drug content to UV light for 72 hours [51].
Method Validation:
- Linearity: Prepare concentrations from 62.5–375 μg/mL, verify correlation coefficient >0.999 [51].
- Precision: Perform six independent assays, RSD should be <2% [51].
- Accuracy: Conduct recovery studies at 50%, 100%, 150% levels, recovery should be 98-102% [51].
- Robustness: Evaluate influence of small variations in flow rate (±0.1 mL/min), mobile phase composition (±2%), buffer pH (±0.2) [51].
Figure 2: Dissolution testing workflow showing critical decision points for method selection and application [50] [46].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Dissolution and Stability Testing
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| Chromatography Columns | Stationary phase for separation | C18, 1.7-1.8μm particles, 50-100mm length [51] [52] |
| Buffer Salts | Mobile phase preparation for pH control | Ammonium acetate, potassium phosphate, sodium acetate [50] [51] |
| Organic Modifiers | Mobile phase for gradient elution | Acetonitrile, methanol (HPLC/UPLC grade) [51] [52] |
| Dissolution Media | Simulate physiological conditions | HCl (pH 1.2), buffers (pH 4-6.8), surfactants [50] [46] |
| Reference Standards | Quantification and method calibration | API with certified purity (>98%) [50] [51] |
| Forced Degradation Reagents | Stress studies for stability indication | HCl, NaOH, H₂O₂, thermal and UV chambers [51] |
| Filters | Sample clarification prior to analysis | 0.20-0.45 μm nylon or PVDF membranes [51] [46] |
Method Selection and Application Framework
The selection between UFLC and spectrophotometric methods depends on multiple factors including study purpose, regulatory requirements, and formulation complexity. UFLC is strongly preferred for stability-indicating methods and complex formulations where separation of degradation products is essential [52] [48]. For dissolution testing of simple formulations without interfering substances, spectrophotometry may provide adequate data with greater efficiency and lower cost [50] [49].
The discrimination power of the dissolution method is paramount, ensuring the method can detect critical differences in formulation performance. As demonstrated with atorvastatin, different pilot batches with varying disintegrant levels showed distinguishable dissolution profiles when using appropriate conditions [50]. This discriminative capacity forms the foundation for establishing meaningful IVIVC, particularly for BCS Class II drugs where dissolution is the rate-limiting step for absorption [50].
For stability-indicating methods, method validation remains a critical regulatory requirement, demonstrating specificity, accuracy, precision, linearity, and robustness according to ICH guidelines [47] [51]. The integration of mass spectrometry with UFLC further enhances degradation product identification, providing comprehensive stability assessment during pharmaceutical development [47] [48].
Quantification of Trace Impurities and Degradation Products
Within pharmaceutical development, the accurate quantification of trace impurities and degradation products is paramount to ensuring drug safety and efficacy. This process is critical for identifying and controlling potentially genotoxic species, which can be present at low levels but pose significant patient risks [53]. The selection of an appropriate analytical technique is therefore a foundational decision for any quality control or stability-testing protocol. This application note details structured methodologies for two principal analytical techniques: ultra-fast Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) and spectrophotometry. Framed within broader research comparing ultra-fast liquid chromatography and spectrophotometric discrimination, this document provides detailed protocols, performance data, and decision frameworks to guide scientists in selecting and implementing the optimal strategy for their specific analytical challenge.
Analytical Techniques: Principles and Comparative Performance
The core of impurity analysis lies in selecting a technique with sufficient selectivity, sensitivity, and specificity. LC-MS/MS and spectrophotometry serve distinct yet sometimes complementary roles.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) combines the physical separation power of liquid chromatography with the exceptional detection and identification capabilities of mass spectrometry. In an LC-MS/MS system, compounds are first separated on a chromatographic column. The eluted compounds are then ionized, and specific precursor ions are selected in the first mass analyzer. These ions are fragmented, and characteristic product ions are detected in the second mass analyzer [54]. This two-stage mass analysis provides a high degree of specificity, effectively isolating the target analyte from complex sample matrices and background noise. Ultra-fast LC-MS/MS leverages sub-2µm particle columns and high-pressure systems to achieve superior speed and resolution [55].
Spectrophotometry, particularly in the CIELAB color space, quantifies color changes by measuring light reflectance or transmittance. It provides objective values for parameters such as L* (lightness), a* (red-green axis), and b* (yellow-blue axis) [56] [18]. While not a separation technique, it is a powerful tool for quantifying gross changes, such as those induced by the thermal degradation of materials, where color change can serve as a proxy for the extent of degradation [56].
The table below summarizes the typical quantitative performance characteristics of these techniques for the analysis of impurities and degradation products.
Table 1: Comparative Performance of LC-MS/MS and Spectrophotometry in Impurity Analysis
| Performance Characteristic | Ultra-Fast LC-MS/MS | Spectrophotometry (CIELAB) |
|---|---|---|
| Primary Application | Identification and quantification of specific chemical impurities [54] [53] | Assessment of gross physical changes (e.g., color) due to degradation [56] |
| Sensitivity | Excellent (e.g., LOD of 0.05 ng/mL for genotoxic impurities) [53] | Limited to macroscopic changes; not for trace chemical analysis [56] |
| Selectivity/Specificity | Very High; uses ion pairs and fragmentation patterns [54] | Low; measures overall color, cannot distinguish between different compounds [18] |
| Linear Range | Wide dynamic range (e.g., 0.2-100 ng/mL with r² ≥ 0.9998) [53] | Demonstrated linear response to physical changes like temperature [56] |
| Key Quantitative Data | Peak area/height of specific ion transitions [54] [53] | L, a, b* coordinates; Whiteness and Yellowness Indexes [56] |
| Discrimination Power | High, capable of distinguishing structurally similar molecules [53] | Moderate, can discriminate between large changes in sample state (AUC 0.9-1.0 for temperature) [56] |
Detailed Experimental Protocols
Protocol for Quantifying Genotoxic Impurities by UHPLC-Orbitrap HRMS
This protocol is adapted from a study determining genotoxic impurities in nifedipine and can be modified for other drug substances [53].
I. Sample Preparation
- Weigh an appropriate amount of the drug substance (e.g., nifedipine).
- Add a suitable volume of methanol to extract the target impurities.
- Dilute the sample to the final required concentration.
II. Instrumentation and Conditions
- System: Ultra-High-Performance Liquid Chromatography coupled with Electrostatic Field Orbitrap High-Resolution Mass Spectrometry (UHPLC-Orbitrap HRMS).
- Column: ACE EXCEL 3 C18-AR (150 mm × 4.6 mm, 3 µm) or equivalent.
- Mobile Phase: Methanol and 0.1% formic acid in water (65:35, v/v), isocratic elution.
- Flow Rate: 0.6 mL/min.
- Column Temperature: 35 °C.
- Injection Volume: As determined during method optimization.
- Mass Spectrometry Conditions:
- Ionization Mode: Electrospray Ionization (ESI), positive mode.
- Scanning Mode: Parallel Reaction Monitoring (PRM).
- Mass Resolution: 35,000 FWHM (Full Width at Half Maximum).
- Sheath/Auxiliary Gas: 60/20 arbitrary units.
- Spray Voltage: 3.5 kV.
- Capillary Temperature: 350 °C.
- Auxiliary Gas Heater Temperature: 400 °C.
- Divert Valve: Set to direct the eluent to waste between 7.5-11.6 min to protect the mass spectrometer from high concentrations of the active pharmaceutical ingredient (API).
III. Quantification and Data Analysis
- Calibration Curve: Prepare and analyze standard solutions of the impurities across the expected concentration range (e.g., 0.2-100 ng/mL). Plot the peak area against the concentration to establish a linear calibration curve.
- Identification: Identify impurities based on the accurate mass of their [M+H]⁺ precursor and fragment ions, and their specific retention times.
- Quantification: Use the external standard method for quantification. The peak areas of the samples are compared against the calibration curve to determine the concentration of each impurity.
Protocol for Assessing Heat-Induced Degradation by Spectrophotometry
This protocol, based on the assessment of heated bone, provides a model for quantifying degradation through color measurement in solid samples [56].
I. Sample Preparation
- Prepare uniform sections of the test material.
- Subject the samples to controlled stress conditions (e.g., heat in a muffle furnace at temperatures such as 200°C, 400°C, 600°C, and 800°C for set durations like 30 or 60 minutes).
- Include an unexposed control group for baseline measurements.
II. Instrumentation and Conditions
- Instrument: Portable contact spectrophotometer.
- Geometry: 8° standard observer.
- Illuminant: D65 (standard daylight).
- Measurement Aperture: 8 mm.
- Calibration: Calibrate the instrument before each measurement session using a white standard tile.
III. Measurement and Data Analysis
- Take multiple measurements (e.g., three) at different sites on both the cortical and medullar zones of the sample to ensure representativeness.
- Record the mean CIELAB values (L, a, b*) and luminance (Y).
- Calculate Whiteness (WI) and Yellowness (YI) Indexes using standard formulas [56].
- Statistical Analysis:
- Use one-way ANOVA with a post-hoc test (e.g., Bonferroni) to compare groups (p < 0.05 considered significant).
- Perform Receiver Operating Characteristic (ROC) analysis to evaluate the accuracy of color parameters in classifying the temperature of exposure, reported as the Area Under the Curve (AUC).
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and solutions required for the experiments described in this note.
Table 2: Key Research Reagent Solutions for Impurity and Degradation Analysis
| Item | Function/Application | Example / Specification |
|---|---|---|
| LC-MS Grade Solvents | Mobile phase preparation; ensures minimal background noise and ion suppression. | Acetonitrile, Methanol, Water (with 0.1% Formic Acid) [57] [53] |
| High-Resolution Mass Spectrometer | Provides accurate mass measurement for definitive identification of impurities. | UHPLC-Orbitrap HRMS System [53] |
| UPLC Column | High-efficiency chromatographic separation with sub-2µm particles. | ACQUITY UPLC BEH C18, 50 x 2.1 mm, 1.7 µm particles [55] |
| Certified Reference Standards | Method development, calibration, and quantification of target impurities. | Impurities 2, 6, and 12 for nifedipine analysis [53] |
| Portable Spectrophotometer | Objective, quantitative measurement of color in solid samples. | Instrument with D65 illuminant and 8° standard observer [56] |
| Forced Degradation Reagents | To intentionally stress the API and generate degradation products for study. | 1 N HCl, 1 N NaOH, H₂O₂ (for acid, base, oxidative stress) [57] |
| Muffle Furnace | Application of controlled thermal stress to samples. | Capable of temperatures up to 800°C with programmable heating rates [56] |
Workflow and Decision Pathway Diagrams
The following diagrams outline the experimental workflow for LC-MS/MS analysis and the logical decision process for selecting the appropriate analytical technique.
Diagram 1: LC-MS/MS Impurity Analysis Workflow
Diagram 2: Analytical Method Selection Guide
The precise quantification of trace impurities and degradation products is a non-negotiable aspect of modern drug development. As demonstrated in this application note, the choice between advanced techniques like ultra-fast LC-MS/MS and simpler spectrophotometric methods is dictated by the specific analytical question. LC-MS/MS is the unequivocal choice for identifying and quantifying specific chemical impurities at trace levels with high specificity, as required by regulatory standards for genotoxic impurities [53]. Conversely, spectrophotometry provides a robust, objective means of quantifying physical degradation phenomena, such as heat-induced color changes, that would otherwise be subject to visual discrimination [56] [18]. By implementing the detailed protocols and decision frameworks provided, researchers can effectively align their analytical strategies with their scientific objectives, thereby ensuring the safety, quality, and stability of pharmaceutical products.
The quantitative analysis of drugs and their metabolites in biological samples like plasma and urine is a cornerstone of pharmaceutical development and therapeutic drug monitoring. This application note details robust methodologies for analyzing such samples, framed within a broader research thesis comparing Ultra-Fast Liquid Chromatography (UFLC) and spectrophotometric techniques. The drive for faster, more efficient, and greener analytical methods has positioned techniques like UPLC-MS/MS and innovative spectrophotometry as critical tools for researchers and drug development professionals. We provide detailed, executable protocols for both technological paths, enabling direct comparison of their capabilities in terms of speed, sensitivity, and suitability for high-throughput environments [19] [58] [59].
Methodological Comparison: UFLC-MS/MS vs. Spectrophotometry
The choice between UFLC-MS/MS and spectrophotometry hinges on the specific requirements of the analysis, including the need for sensitivity, specificity, throughput, and the complexity of the biological matrix. The table below summarizes the core characteristics of each approach for bioanalysis.
Table 1: Comparison of UFLC-MS/MS and Spectrophotometry for Bioanalysis
| Feature | UFLC-MS/MS | Spectrophotometry |
|---|---|---|
| Key Principle | Chromatographic separation followed by mass-based detection [19] | Measurement of light absorption by analytes at specific wavelengths [39] |
| Typical Analysis Time | ~1.5 - 4.0 minutes per sample [19] [58] | Rapid (minutes, including sample prep) [37] |
| Sensitivity | High (e.g., LLOQ of 0.2 ng/mL for donepezil in plasma) [19] | Moderate (e.g., LOD of 0.30 µg/mL for Dronedarone HCl) [60] |
| Specificity | Very High (separation + mass detection) [61] | Low to Moderate (susceptible to matrix interference) [39] |
| Sample Throughput | High (with fast gradients and automation) [30] | Very High (suitable for batch analysis) [37] |
| Data Richness | High (multiplexed quantification, structural info) [62] | Low (single-analyte focus typically) |
| Greenness (AGREE Score Example) | 0.77 (for a revumenib assay) [58] | >0.77 (often higher due to less solvent use) [37] |
| Ideal Application | Bioequivalence studies, metabolite identification, multi-analyte panels [19] [61] | Quality control of formulations, analysis of single drugs in simple matrices [37] [39] |
Experimental Protocols
Protocol 1: Ultra-Fast LC-MS/MS for Drug Quantification in Plasma
This protocol for the determination of donepezil in human plasma exemplifies a robust UFLC-MS/MS approach suitable for supporting bioequivalence studies [19].
Materials and Reagents
- Analytical Standard: Donepezil and deuterium-labeled Internal Standard (Donepezil-d5) [19].
- Mobile Phase A: 0.1% Formic acid in water [19].
- Mobile Phase B: 0.1% Formic acid in acetonitrile [19].
- Biological Matrix: Control human plasma with K₂EDTA as an anticoagulant [19].
- Precipitation Solvent: HPLC-grade methanol [19].
Instrumentation and Conditions
- Chromatography: UHPLC system with a monolithic column (e.g., Chromolith High Resolution RP-18e, 50 × 4.6 mm) [19].
- Mass Spectrometry: Triple quadrupole mass spectrometer with electrospray ionization (ESI) in positive mode [19].
- MRM Transitions: m/z 380 → 91 for donepezil; m/z 385 → 96 for the IS [19].
- Gradient & Flow: Multi-stage flow rate, starting at 3 mL/min and reducing to 1.2 mL/min for the analytical separation. Total run time: 1.5 minutes [19].
- Injection Volume: 5 µL [19].
Sample Preparation Procedure
- Thaw frozen plasma samples at room temperature.
- Pipette 200 µL of plasma into a 96-well plate.
- Add 50 µL of the internal standard working solution.
- Precipitate proteins by adding 500 µL of methanol.
- Vortex mix thoroughly and centrifuge at 3,500 × g for 5 minutes at 10°C.
- Transfer 200 µL of the supernatant to a new plate and add 400 µL of water.
- Inject a 5 µL aliquot into the LC-MS/MS system [19].
Method Validation
The method was validated per FDA/ICH guidelines, demonstrating [19]:
- Linearity: 0.2 - 50 ng/mL (r > 0.995).
- Precision & Accuracy: Intra- and inter-day accuracy and precision within ±15%.
- Recovery: Consistent extraction efficiency across the calibration range.
- Specificity: No interference from plasma matrix components.
Protocol 2: Green Spectrophotometric Analysis for Formulations
This protocol outlines a green, third-derivative spectrophotometry (D³) method for resolving and quantifying drug mixtures in formulations, such as Terbinafine HCl (TFH) and Ketoconazole (KTZ), without prior separation [37].
Materials and Reagents
- Analytical Standards: TFH and KTZ reference standards [37].
- Solvent: Methanol and distilled water [37].
Instrumentation and Conditions
- Instrument: Double-beam UV-Vis Spectrophotometer (e.g., Shimadzu UV-1900i) [37].
- Wavelengths: 214.7 nm for TFH and 208.6 nm for KTZ (from third-derivative spectra) [37].
- Spectral Processing Parameters: Scaling factor = 10, Δλ = 8 nm [37].
Sample Preparation and Calibration
- Prepare stock solutions of TFH and KTZ at 1.0 mg/mL in methanol.
- Dilute further with distilled water to create working solutions of 100 µg/mL.
- Prepare calibration standards in the range of 0.6–12.0 µg/mL for TFH and 1.0–10.0 µg/mL for KTZ in distilled water [37].
- For tablet analysis, weigh and powder tablets. Dissolve an equivalent weight of powder in methanol, sonicate, filter, and dilute to volume with distilled water to a concentration within the working range [37].
Measurement and Analysis
- Record the zero-order absorption spectra of standards and samples against a distilled water blank.
- Obtain the third-derivative spectra (D³) using the instrument's software.
- Measure the amplitudes of the D³ spectra at 214.7 nm for TFH and 208.6 nm for KTZ.
- Construct calibration curves by plotting the D³ amplitude versus concentration and determine the regression equations [37].
Table 2: Validation Parameters for Spectrophotometric Methods (Examples)
| Validation Parameter | Terbinafine HCl (D³ Method) [37] | Repaglinide (UV at 241 nm) [39] |
|---|---|---|
| Linearity Range (µg/mL) | 0.6 - 12.0 | 5 - 30 |
| Correlation Coefficient (r²) | > 0.9992 (reported for similar methods) [60] | > 0.999 |
| LOD (µg/mL) | ~0.30 (reported for similar methods) [60] | Not Specified |
| LOQ (µg/mL) | 1.0 (reported for similar methods) [60] | Not Specified |
| Precision (% RSD) | < 2.0 | < 1.50 |
| Accuracy (% Recovery) | 98 - 102 | 99.63 - 100.45 |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful bioanalysis requires carefully selected, high-purity materials. The following table lists key reagents and their critical functions in the workflows described.
Table 3: Essential Research Reagent Solutions for Bioanalysis
| Item | Function & Application |
|---|---|
| Analytical Reference Standards | Provides a pure benchmark for identifying the analyte and constructing calibration curves for quantitative analysis [19] [37]. |
| Deuterated Internal Standards (e.g., Donepezil-d5) | Compensates for variability in sample preparation and ionization efficiency in LC-MS/MS, significantly improving accuracy and precision [19]. |
| HPLC-MS Grade Solvents (ACN, MeOH, Water) | Ensure low UV background, minimal ion suppression, and prevent system contamination, which is crucial for sensitivity and reproducibility [19] [58]. |
| Acid Additives (Formic Acid, Ammonium Formate) | Volatile mobile phase modifiers that enhance analyte protonation and ionization efficiency in ESI-MS, improving signal intensity [19] [58]. |
| Protein Precipitation Reagents (e.g., Methanol, ACN) | Remove proteins from biological samples like plasma and urine, minimizing matrix effects and protecting the analytical column [19]. |
| Human Liver Microsomes (HLMs) | An in vitro system used to study a drug's metabolic stability and identify its metabolites during early development [58]. |
UFLC-MS/MS and spectrophotometry serve distinct but vital roles in the modern analytical laboratory. UFLC-MS/MS is the unequivocal choice for high-sensitivity, multi-analyte quantification in complex biological matrices like plasma and urine, particularly when supporting regulatory submissions. Meanwhile, advanced, green spectrophotometric methods offer a rapid, cost-effective, and reliable solution for analyzing formulations and less complex samples. The protocols and data presented herein provide a framework for researchers to select and implement the optimal methodology based on their specific analytical target profile, thereby enhancing efficiency and decision-making in drug development.
In the evolving landscape of pharmaceutical analysis, the discrimination between ultra-fast liquid chromatography (UFLC) and spectrophotometric methods represents a significant research frontier. While UFLC and UHPLC-MS/MS offer superior separation capabilities and sensitivity for complex matrices [63], spectrophotometry remains indispensable for its simplicity, cost-effectiveness, and rapid analysis [64] [65]. The strategic selection of reagents—complexing agents, pH indicators, and diazotization reagents—enhances the capability of spectrophotometric methods to achieve the selectivity and sensitivity required for modern drug development [64]. This article provides detailed application notes and protocols to guide researchers in leveraging these reagents effectively, framing their use within the comparative context of advanced chromatographic techniques.
Core Principles and Comparative Context
Spectrophotometry in the Modern Laboratory
The principle of spectrophotometry is governed by the Beer-Lambert Law (A = εcl), which establishes a linear relationship between absorbance (A) and the concentration (c) of an analyte [66] [64] [65]. Its advantages include high sensitivity, accuracy, and non-destructive analysis, making it suitable for routine quality control, dissolution studies, and stability testing where ultra-fast LC may be unnecessarily complex or costly [64] [39] [65].
Strategic Role of Reagents
Many pharmaceutical compounds lack inherent chromophores, necessitating derivatization to produce measurable signals. The reagents discussed herein facilitate this:
- Complexing Agents: Form stable, colored complexes with metal ions or specific functional groups.
- pH Indicators: Exploit acid-base equilibria to induce measurable color changes.
- Diazotization Reagents: Convert primary aromatic amines into highly colored azo dyes.
The judicious application of these reagents allows spectrophotometry to maintain a competitive position in the analytical toolkit, particularly for applications where speed and cost are paramount.
Complexing Agents
Principles and Applications
Complexing agents react with analytes to form stable, colored complexes, thereby enhancing both the sensitivity and selectivity of spectrophotometric methods [64]. They are particularly crucial for quantifying metal ions or drugs that can coordinate with metal centers [64].
Table 1: Characteristics of Selected Complexing Agents
| Complexing Agent | Target Analyte | Key Spectral Properties | Application Notes | Source |
|---|---|---|---|---|
| Desferrioxamine B (DFO) | Iron (Fe³⁺) | λmax = ~430 nm; ε is high | Forms a stable 1:1 complex; wide pH range (3.5-8); determines total iron. | [66] |
| Calmagite | Calcium (Ca²⁺) | Absorbance decrease at ~610 nm | Used in alkaline conditions (pH ~11); rapid complexation. | [67] |
| Ferric Chloride | Phenolic compounds (e.g., Paracetamol) | Varies by complex | Used for drugs with phenolic functional groups. | [64] |
Detailed Protocol: Determination of Total Iron Using Desferrioxamine B (DFO)
This protocol is adapted from a method for determining iron in natural waters and biological materials [66].
1. Principle: DFO forms an intensely red-colored, stable 1:1 complex with Fe³⁺. The complex is so stable that any Fe²⁺ present is oxidized and complexed, allowing for the determination of total iron content [66].
2. Research Reagent Solutions:
- Desferrioxamine B Solution: 0.008 M DFO in water.
- Iron Standard Stock Solution: 1000 mg/L, prepared from Fe(NO₃)₃ in 0.3 M nitric acid.
- Sodium Hydroxide Solution: 0.1 M, for pH adjustment.
- Buffer Solution: Not strictly required, as the complex is stable over a wide pH range.
3. Procedure: 1. Preparation of Calibration Standards: Pipette increasing amounts of the iron standard stock solution into a series of 25 mL volumetric flasks. 2. Complex Formation: To each flask, add 5.0 mL of the 0.008 M DFO solution. 3. pH Adjustment: Add a sufficient volume of 0.1 M NaOH to neutralize the acid from the standard and the H⁺ released during complexation, bringing the final pH to between 6.8 and 7.1. 4. Dilution: Dilute the contents to the mark with deionized water and mix thoroughly. 5. Spectrophotometric Measurement: Measure the absorbance of each solution against a reagent blank at the absorption maximum (approximately 430 nm) using a 1 cm pathlength cell. 6. Calibration and Analysis: Construct a calibration curve by plotting absorbance versus the known iron concentration. The data should conform to the Beer-Lambert law. Determine the concentration of unknown samples from this curve.
4. Critical Notes:
- The [FeLH]+ complex is stable in the pH range of 3.5 to 8, making the method robust against minor pH variations [66].
- The complex formation is rapid at pH values above 3 [66].
pH Indicators
Principles and Applications
pH indicators are weak acids or bases that change color depending on the protonation state, which is controlled by the solution's pH [64]. This property is harnessed to analyze the acid-base character of pharmaceutical compounds, which can influence drug stability, solubility, and bioavailability [64].
Table 2: Characteristics of Selected pH Indicators
| pH Indicator | Color Change & pH Range | Analytical Application | Application Notes | Source |
|---|---|---|---|---|
| Bromocresol Green | Yellow (pH 3.8) to Blue (pH 5.4) | Assay of weak acids | Forms an ion-pair complex with basic drugs; extractable into organic solvents. | [64] |
| Phenolphthalein | Colorless (pH < 8.3) to Pink/Fuchsia (pH > 10.0) | Analysis of base-forming drugs | Classic acid-base titrimetric endpoint indicator. | [64] |
Detailed Protocol: Analysis of a Basic Drug Using Bromocresol Green
This protocol outlines a general method for assaying basic drugs via ion-pair formation.
1. Principle: In an aqueous buffer, the protonated form of a basic drug (D+) forms an ion-pair with the anionic form of the indicator (In−). This ion-pair complex is often extractable into an organic solvent, allowing for its concentration and spectrophotometric measurement [64].
2. Research Reagent Solutions:
- Bromocresol Green Solution: 0.05% w/v in distilled water.
- Buffer Solution: Phosphate or acetate buffer, typically at pH 3.5-4.5, to ensure drug protonation and indicator anionic form.
- Organic Solvent: Chloroform or dichloromethane, for extraction.
3. Procedure: 1. Sample Preparation: Dissolve or dilute the drug sample in the aqueous buffer solution. 2. Ion-Pair Formation: Add a known volume of the bromocresol green solution to the drug solution and mix. 3. Extraction: Transfer the mixture to a separatory funnel and extract the colored ion-pair complex with a known volume of organic solvent (e.g., chloroform). 4. Separation: Allow the layers to separate completely. Collect the organic layer, which contains the colored complex. 5. Spectrophotometric Measurement: Measure the absorbance of the organic extract against an organic solvent blank at the wavelength of maximum absorption (typically ~415-420 nm). 6. Calibration and Analysis: Construct a calibration curve using standard solutions of the drug treated identically.
4. Critical Notes:
- The pH of the buffer is critical for quantitative ion-pair formation and extraction.
- The organic solvent must be of high purity to avoid interfering absorbance.
Diazotization Reagents
Principles and Applications
Diazotization involves the conversion of a primary aromatic amine into a diazonium salt using nitrous acid (generated in situ from sodium nitrite and acid). This diazonium salt is then coupled with a suitable agent to form a highly colored azo dye [64] [68]. This method is exceptionally sensitive for drugs containing primary aromatic amine groups.
Table 3: Reagents for Diazotization and Coupling
| Reagent | Type | Function | Application Example | Source |
|---|---|---|---|---|
| Sodium Nitrite & HCl | Diazotization | Generates nitrous acid to form diazonium salt | Used in analysis of sulfonamides and other aryl amines. | [64] [68] |
| N-(1-Naphthyl)ethylenediamine (NEDH) | Coupling Agent | Couples with diazonium salt to form azo dye | Official method for nitrite and sulfonamide analysis. | [64] [68] |
| Ethyl Acetoacetate (EAA) | Coupling Agent | Alternative coupling agent | Used with p-nitroaniline or sulfanilamide for nitrite determination. | [68] |
Detailed Protocol: Determination of Nitrite Using a Diazotization-Coupling Reaction
This protocol is based on a method for determining nitrite in water and soil samples using ethyl acetoacetate (EAA) as a coupling agent [68].
1. Principle: Nitrite reacts with a primary aromatic amine (like p-nitroaniline or sulfanilamide) in an acidic medium to form a diazonium salt. This salt subsequently couples with EAA in an alkaline medium to produce a water-soluble, colored azo dye suitable for direct spectrophotometric measurement [68].
2. Research Reagent Solutions:
- p-Nitroaniline (PNA) Solution: 0.2% w/v in 1 M HCl.
- Sodium Nitrite Solution: Standard solution of known concentration for calibration.
- Ethyl Acetoacetate (EAA) Solution: 2% v/v in ethanol.
- Sodium Hydroxide Solution: 2 M, for alkalinity.
3. Procedure: 1. Diazotization: To a known volume of sample or standard nitrite solution in a volumetric flask, add 1 mL of the PNA solution. Mix and allow to stand for a few minutes for the diazotization reaction to complete. 2. Coupling: Add 1 mL of the EAA solution, followed by 2 mL of the 2 M sodium hydroxide solution. The solution will develop a color. 3. Dilution and Stability: Make up to the mark with distilled water, mix well, and allow the color to develop fully. The developed color is stable for at least 2 hours. 4. Spectrophotometric Measurement: Measure the absorbance against a reagent blank at 507 nm if PNA was used. 5. Calibration and Analysis: Construct a calibration curve using nitrite standards and determine the concentration of the unknown.
4. Critical Notes:
- The method is characterized by rapid color development, excellent reproducibility, and relative independence from pH and temperature effects [68].
- Reagents are inexpensive and readily available.
Method Validation and Comparative Analysis with UFLC
Validation of Spectrophotometric Methods
For any analytical method, validation is crucial. According to ICH guidelines, key parameters for spectrophotometric methods include [39] [69]:
- Linearity: Demonstrated over a defined concentration range (e.g., 5-30 μg/mL for Repaglinide) with a regression coefficient (r²) > 0.999 [39].
- Precision: Both repeatability (intra-day) and intermediate precision (inter-day) should yield %R.S.D. values < 1.5% [39].
- Accuracy: Established through recovery studies, with mean recoveries ideally between 99-101% [39].
- LOD and LOQ: Calculated based on the standard deviation of the response and the slope of the calibration curve [39].
Strategic Discrimination: Spectrophotometry vs. UFLC/UHPLC
The choice between spectrophotometry and UFLC is not a matter of superiority but of strategic application. The following table outlines the key discriminators.
Table 4: Spectrophotometry vs. UFLC/UHPLC for Pharmaceutical Analysis
| Parameter | Spectrophotometry (with Derivatization) | UFLC / UHPLC-MS/MS |
|---|---|---|
| Cost and Complexity | Low cost, simple instrumentation and operation [64] [65]. | High cost, complex instrumentation and operation [63]. |
| Analysis Speed | Very fast for single-analyte tests [65]. | Faster separation times but often longer overall method run times [70] [63]. |
| Selectivity | Good for single analytes; can suffer from interference in complex matrices [64]. | Excellent selectivity, especially when hyphenated with MS/MS [70] [63]. |
| Sensitivity | Sufficient for most dosage form assays (e.g., LoQ in μg/mL range) [39]. | Superior sensitivity (LoQ in ng/mL or pg/mL), essential for bioanalysis and trace impurities [63]. |
| Multi-analyte Capability | Limited; typically for one analyte at a time. | High; capable of simultaneous multi-analyte and multi-impurity profiling [70] [63]. |
| Ideal Application | Routine quality control of APIs in bulk and formulations, dissolution testing [64] [69]. | Bioanalysis, metabolomics, complex impurity profiling, stability-indicating methods [63]. |
The strategic selection of reagents—complexing agents, pH indicators, and diazotization systems—empowers spectrophotometry to remain a vital and robust technique in the pharmaceutical scientist's arsenal. While UFLC and UHPLC-MS/MS are unequivocally superior for the analysis of complex mixtures, trace-level analytes, and demanding stability-indicating assays [70] [63], spectrophotometry offers an unbeatable combination of speed, simplicity, and cost-effectiveness for a well-defined set of applications [64] [65]. A thorough understanding of the principles and protocols provided in these application notes will enable researchers and drug development professionals to make informed, discriminatory choices between these powerful analytical techniques, optimizing resource allocation and efficiency in the drug development pipeline.
Recent Column Innovations for Small Molecules and Biomolecules
Application Note: Advanced Stationary Phases for Ultrafast Separations
The pursuit of higher analytical throughput in pharmaceutical development has driven significant innovation in liquid chromatography (LC) column technology. This application note details recent column advancements that enable ultra-fast, high-resolution separations of both small molecules and biomolecules, contrasting these modern approaches with traditional spectrophotometric methods. While spectrophotometry remains valued for its simplicity and cost-effectiveness in drug assay and impurity profiling [64], its limitations in specificity and sensitivity for complex mixtures have accelerated the adoption of advanced LC techniques [71]. The innovations highlighted herein focus on enhancing separation efficiency, improving analyte recovery, and expanding applicability across diverse compound classes.
Recent Column Innovations
The table below summarizes key recently commercialized LC columns designed to address specific analytical challenges in pharmaceutical analysis.
Table 1: Recent Innovations in Liquid Chromatography Columns for Pharmaceutical Analysis
| Product Name | Manufacturer | Stationary Phase Characteristics | Key Features and Benefits | Target Applications |
|---|---|---|---|---|
| Halo Inert [72] | Advanced Materials Technology | Superficially porous particles with passivated hardware | Metal-free flow path; enhances peak shape and recovery for metal-sensitive analytes | Phosphorylated compounds, metal-chelating molecules |
| Evosphere C18/AR [72] | Fortis Technologies Ltd. | Monodisperse fully porous particles with C18/aromatic ligands | Oligonucleotide separation without ion-pairing reagents; higher efficiency | Oligonucleotide analysis, biopharmaceuticals |
| Ascentis Express BIOshell A160 Peptide PCS-C18 [72] | Merck Life Sciences | Superficially porous particles with positively charged surface | Enhanced peak shapes for basic compounds and peptides; high throughput | Peptide mapping, pharmaceutical analysis |
| Raptor Inert HPLC Columns [72] | Restek Corporation | Superficially porous silica particles (1.8/2.7 μm) with various functional groups | Inert hardware improves response for metal-sensitive polar compounds | Analysis of chelating compounds, polar molecules |
| Aurashell Biphenyl [72] | Horizon Chromatography | Superficially porous particles with biphenyl functional groups | π-π interactions, dipole, steric effects; enhanced polar compound retention | Metabolomics, isomer separations, polar aromatics |
| Halo 90 Å PCS Phenyl-Hexyl [72] | Advanced Materials Technology | Superficially porous particles with phenyl-hexyl group | Alternative selectivity to C18; improved peak shape for basic compounds | Mass spectrometry applications, method development |
Quantitative Performance Comparison
The following table compares the quantitative performance of a documented ultrafast monolithic column method against a conventional high-performance LC-MS/MS method for the analysis of donepezil in human plasma, demonstrating the efficiency gains of modern approaches.
Table 2: Quantitative Method Performance: Ultrafast Monolithic vs. Conventional LC-MS/MS [19]
| Parameter | Ultrafast Monolithic Method | Conventional Method |
|---|---|---|
| Stationary Phase | Chromolith High Resolution RP-18e | InfinityLab Poroshell 120 EC-C18 |
| Column Dimensions | 50 × 4.6 mm | 2.1 × 50 mm, 2.7-Micron |
| Total Run Time | 1.5 minutes | 4.0 minutes |
| Linear Dynamic Range | 0.2–50 ng/mL | 0.2–50 ng/mL |
| Intra-/Inter-day Accuracy & Precision | Within 15% | Within 15% |
| Sample Preparation | Protein precipitation | Protein precipitation |
| Key Advantage | Ultra-fast analysis with sustainable performance | Established, robust methodology |
Experimental Protocols
Protocol 1: Ultrafast LC-MS/MS Analysis of Donepezil Using a Monolithic Column
Application: Bioequivalence study support for donepezil hydrochloride formulations [19].
Principle: This protocol utilizes a monolithic stationary phase, which possesses a biporous structure (macropores for high flow and mesopores for high surface area) enabling separations at high flow rates with low backpressure. Multi-stage flow rate programming combined with a diverter valve ensures optimal ionization efficiency and minimizes source contamination in the mass spectrometer.
Materials and Reagents:
- Analytes: Donepezil and Donepezil-d5 (Internal Standard, IS)
- Mobile Phase A: 0.1% formic acid in water (v/v)
- Mobile Phase B: 0.1% formic acid in acetonitrile (v/v)
- Column: Chromolith High Resolution RP-18e monolithic column (50 × 4.6 mm)
- System: LC-MS/MS with electrospray ionization (ESI) and a triple quadrupole mass spectrometer
Instrumental Conditions:
- Mass Spectrometer: Positive ion mode; Multiple Reaction Monitoring (MRM)
- Donepezil transition: m/z 380 → 91
- IS transition: m/z 385 → 96
- Column Temperature: Room temperature
- Injection Volume: 5 μL
- Gradient Program & Multi-stage Flow:
- 0.0 - 0.1 min: 75% A, 25% B at 3.0 mL/min
- 0.1 - 0.6 min: Linear to 40% A, 60% B, flow reduced to 1.2 mL/min
- 0.6 - 1.1 min: Linear to 20% A, 80% B at 1.2 mL/min
- 1.1 - 1.2 min: Hold at 20% A, 80% B at 1.2 mL/min
- 1.2 - 1.5 min: Re-equilibrate at 75% A, 25% B, flow increased to 3.0 mL/min
- Diverter Valve: Switches to waste from 0.0-0.6 min and after 1.1 min; directs to MS from 0.6-1.1 min.
Sample Preparation Procedure:
- Thaw frozen human plasma samples at room temperature.
- Aliquot 200 μL of plasma into a 96-well polypropylene plate.
- Add 50 μL of IS working solution to each sample.
- Precipitate proteins by adding 500 μL of methanol.
- Vortex mix the samples vigorously for several minutes.
- Centrifuge at 3,500 × g for 5 minutes at 10°C.
- Transfer 200 μL of the supernatant to a new 96-well plate.
- Add 400 μL of water to the extract and mix.
- Inject a 5 μL aliquot into the LC-MS/MS system.
Validation Parameters:
- Linearity: Eight-point calibration curve (0.2-50 ng/mL) with 1/χ² weighting.
- Sensitivity: LLOQ of 0.2 ng/mL with signal-to-noise ≥5 and accuracy of 80-120%.
- Precision & Accuracy: Intra- and inter-day values within 15% for all QC levels.
- Specificity: No interference from blank plasma in six different lots.
Diagram 1: Ultrafast LC-MS/MS Workflow. The method uses multi-stage flow rates and diverter valve switching to optimize analysis time and MS performance. [19]
Protocol 2: High-Resolution Polysorbate 20 Characterization by HPLC-CAD and UHPLC-Q-TOF-MS
Application: Discrimination of polysorbate 20 (PS20) samples from various origins and degradation states [73].
Principle: This two-dimensional analytical protocol combines high-performance liquid chromatography with charged aerosol detection (HPLC-CAD) for separation and quantification of key PS20 components, with ultra-high-performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS) for precise characterization and database expansion. The identical gradient in both systems ensures correlative data.
Materials and Reagents:
- Samples: PS20 from different sources and grades
- Columns: As specified in the source method for HPLC and UHPLC separation
- Mobile Phases: Appropriate MS-compatible solvents (e.g., water, acetonitrile with modifiers)
- Systems:
- HPLC system coupled to a Charged Aerosol Detector (CAD)
- UHPLC system coupled to a Quadrupole Time-of-Flight Mass Spectrometer (Q-TOF-MS)
Experimental Procedure:
- HPLC-CAD Analysis:
- Separate the PS20 sample using a developed single-dimensional HPLC method.
- Use the CAD for detection, which provides a uniform response factor for non-volatile and semi-volatile analytes independent of chemical structure.
- Generate a chromatographic fingerprint showing 18 key component peaks with multiple esters.
UHPLC-Q-TOF-MS Characterization:
- Analyze the same PS20 samples using a UHPLC method with an identical gradient as the HPLC-CAD analysis.
- Perform high-resolution mass spectrometry in positive/negative ionization modes.
- Identify compounds based on accurate mass and fragmentation patterns.
Database and Library Construction:
- Expand the polysorbate compound database by integrating HPLC retention time and Q-TOF-MS spectral data.
- The method expanded the compound database over 7-fold compared to commercial libraries, identifying 1329 to 1511 compounds in different PS20 batches.
Data Integration and Discrimination:
- Correlate HPLC-CAD fingerprint differences with compositional data from UHPLC-Q-TOF-MS.
- Use this combined data to distinguish PS20 samples from various origins and assess the impact of different degradation conditions (e.g., oxidative, thermal stress) on component profiles.
Diagram 2: PS20 Analysis Workflow. Integrated HPLC-CAD and UHPLC-Q-TOF-MS approach enables comprehensive characterization and sample discrimination. [73]
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Advanced Chromatographic Analysis
| Tool/Reagent | Function and Application | Key Characteristics |
|---|---|---|
| Monodisperse Fully Porous Particles (MFPP) [72] | Stationary phase base for high-efficiency columns (e.g., Evosphere series). | Uniform particle size distribution for superior efficiency and resolution compared to conventional polydisperse particles. |
| Inert Column Hardware [72] | Passivated surfaces (e.g., Halo Inert, Raptor Inert) to minimize metal-analyte interactions. | Prevents adsorption and degradation of metal-sensitive analytes like phosphorylated compounds, improving peak shape and recovery. |
| Charged Aerosol Detector (CAD) [73] | Universal detector for non-volatile and semi-volatile analytes, used in complex mixture analysis (e.g., PS20). | Provides uniform response independent of chemical structure, ideal for quantifying compounds without chromophores. |
| Deuterated Internal Standards [19] | Internal standard for LC-MS/MS quantification (e.g., Donepezil-d5 for Donepezil assay). | Corrects for variability in sample preparation, injection, and ionization; ensures assay accuracy and precision. |
| Superficially Porous Particles [72] | Stationary phase particles (e.g., in Halo, Raptor columns) with a solid core and porous shell. | Enable fast mass transfer, yielding high efficiency at lower backpressures compared to fully porous particles. |
| Shielded Hydrophobic Phase (SHP) Column [74] | For direct injection of biological samples by retaining small molecules while excluding proteins. | Hydrophilic network shields hydrophobic pockets; prevents protein binding but requires ion mobility MS to address polymer bleed. |
Solving Common Problems: Optimization and Troubleshooting Strategies
Diagnosing and Resolving UFLC Pressure Issues and Peak Problems
Ultra-Fast Liquid Chromatography (UFLC) represents a significant advancement in chromatographic science, offering superior speed and resolution compared to conventional HPLC. However, the high operating pressures and complex method parameters inherent to UFLC systems can lead to specific challenges, including pressure abnormalities and peak distortions. For researchers engaged in comparative studies between UFLC and spectrophotometric methods, such as those in drug development, maintaining system integrity is paramount. Spectrophotometric techniques, while economical and simple, often struggle with resolving overlapping spectral bands of analytes and interferences, a limitation that derivative spectrophotometry can only partially overcome [75]. UFLC addresses this fundamental constraint by providing physical separation of components, making the reliable diagnosis and resolution of its operational issues critical for generating high-quality, reproducible data that can be confidently compared across analytical platforms.
This document provides a structured framework for diagnosing and resolving the most common UFLC problems, ensuring data integrity for your research.
Troubleshooting Pressure Issues
Pressure-related problems are among the most frequent issues encountered in UFLC. Due to the higher operating pressures in UFLC systems (often extending into the UHPLC range of 4000–15,000 psi), a small blockage can cause a more dramatic pressure spike than in standard HPLC [76]. Systematic diagnosis is key to a swift resolution.
Diagnosis and Resolution Protocols
The flowchart below provides a systematic pathway for diagnosing the root cause of pressure problems in your UFLC system.
Quantitative Pressure Reference Guide
Familiarity with normal and abnormal pressure ranges helps in early problem detection. The following table summarizes key pressure-related symptoms, their common causes, and specific solutions.
Table 1: UFLC Pressure Issue Troubleshooting Guide
| Symptom | Common Causes | Resolution Protocols |
|---|---|---|
| Sudden Pressure Spike [77] [76] | - Column blockage (particulates, sample precipitate) [78].- Clogged inlet frit or guard column.- Buffer salt precipitation in pump or tubing.- Mobile phase viscosity too high. | 1. Column Reversal: Reverse-flush the column with a strong solvent (e.g., 50:50 methanol/water) if permitted by the manufacturer [76].2. Frit Cleaning: Replace or sonicate the column inlet frit to remove debris.3. System Flushing: Flush the entire system, including the pump, with a 50:50 water/methanol mixture to dissolve salt crystals [76] [78].4. Solvent Adjustment: Use lower flow rates or a less viscous mobile phase composition. |
| Gradual Pressure Increase [79] | - Guard column exhaustion.- Contaminant buildup on the analytical column head.- Microbial growth in mobile phase or buffer. | 1. Guard Column Replacement: Replace the guard cartridge regularly, ensuring it matches the analytical column phase [79].2. Column Cleaning: Flush the analytical column following the manufacturer's recommended protocol, often using a gradient of strong solvents.3. Mobile Phase Management: Prepare fresh, filtered mobile phase daily; use HPLC-grade solvents and water. |
| Low Pressure or Fluctuating Pressure [77] [78] | - Leak in tubing, fittings, or pump seals.- Air bubbles in the pump head.- Worn pump seals or check valves.- Solvent reservoir running low. | 1. Leak Check: Inspect all fittings and unions for mobile phase; tighten or replace as needed [76].2. Air Purge: Purge the pump thoroughly with degassed mobile phase to remove trapped air [78].3. Preventive Maintenance: Replace pump seals and perform routine check valve cleaning every 6-12 months, depending on usage [76]. |
Troubleshooting Peak Problems
Peak shape and retention anomalies directly impact data quality, resolution, and quantitative accuracy. Understanding their origins is essential, especially when comparing UFLC data to spectrophotometric results, where such physical separation does not exist.
Diagnosis and Resolution Protocols
The following workflow guides you through the logical steps of investigating common peak issues.
Quantitative Peak Problem Reference Guide
Table 2: UFLC Peak Shape and Retention Issue Troubleshooting Guide
| Symptom | Common Causes | Resolution Protocols |
|---|---|---|
| Peak Tailing [77] [80] [79] | - Secondary Interactions: Analyte interaction with active silanol groups on the stationary phase.- Column Overload: Too much analyte mass or volume [77].- Void in Column Inlet: Physical degradation of the column bed.- Contamination: Sample matrix components or precipitates on the column head. | 1. Mobile Phase Buffering: Add buffer (e.g., ammonium formate with formic acid) to the mobile phase to block active silanol sites [79].2. Reduce Sample Load: Dilute the sample or decrease the injection volume. For a 2.1 mm ID column, typical injection volumes are 1-3 µL [79].3. Column Inspection: Examine the column for voids; replace if necessary. Use a guard column to prevent contamination. |
| Peak Fronting [77] [80] | - Column Overload (mass or volume) [77].- Sample Solvent Too Strong: Injection solvent is stronger than the mobile phase.- Channeled Column: Physical damage to the column packing. | 1. Optimize Solvent Strength: Dissolve the sample in the starting mobile phase composition or a weaker solvent [79].2. Reduce Sample Load: Dilute the sample or reduce the injection volume.3. Column Replacement: Replace the column if the packing is physically damaged. |
| Retention Time Shifts [77] [78] | - Mobile Phase Composition Change: Evaporation, incorrect preparation, or pump malfunction.- Column Temperature Fluctuation: Unstable oven temperature.- Column Degradation: Stationary phase aging or damage. | 1. Mobile Phase Preparation: Prepare fresh mobile phase consistently and keep reservoirs capped.2. System Verification: Verify column oven temperature stability and pump flow rate accuracy [77].3. Column Care: Equilibrate column thoroughly; replace aged column. |
| Ghost Peaks [77] | - Carryover: Incomplete cleaning of the autosampler.- Contaminants: In mobile phase, sample vials, or from system components (e.g., pump seals).- Column Bleed: Decomposition of the stationary phase. | 1. Run Blanks: Perform blank injections to profile ghost peaks.2. Thorough Cleaning: Clean the autosampler (needle, loop, needle seat) and use high-purity solvents [77].3. System Maintenance: Replace worn seals and tubing; if using MS detection, ensure solvents are LC-MS grade. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for UFLC Maintenance and Troubleshooting
| Item | Function / Purpose | Notes |
|---|---|---|
| HPLC/MS Grade Solvents | High-purity solvents minimize baseline noise and ghost peaks, crucial for sensitive detection. | Essential for MS-compatible methods to avoid ion suppression and contamination [79]. |
| Ammonium Formate/Acetate | Used to prepare buffered mobile phases that suppress silanol interactions and stabilize ionization. | Preferable to non-volatile salts for LC-MS applications [79]. |
| Guard Columns | Small cartridge placed before the analytical column to trap contaminants and particulates. | Extends analytical column lifetime; should match the stationary phase of the analytical column [79]. |
| In-line Filters | Placed between the injector and guard column to capture particulate matter. | Protects the guard and analytical columns from blockages. |
| Column Cleaning Solvents | Strong solvents (e.g., high % acetonitrile or methanol, isopropanol) for flushing contaminants. | Follow column manufacturer's pH and pressure limits during cleaning. |
| Certified Standard Mixture | A sample with known peak shape and retention used for system performance checks. | Critical for diagnosing whether an issue is method- or system-related [79]. |
Effective troubleshooting of UFLC systems is a systematic process that relies on understanding symptom-cause relationships and methodically testing potential solutions. Pressure and peak shape issues can significantly compromise the high-quality data that UFLC is capable of producing, especially when these results form the basis for comparison with other analytical techniques like spectrophotometry. By establishing a baseline of normal system performance, adhering to a structured diagnostic workflow, and implementing proactive maintenance protocols, researchers can ensure the reliability and reproducibility of their chromatographic data, thereby strengthening the conclusions drawn from their research.
In the rigorous comparative analysis between ultra-fast liquid chromatography (UFLC) and spectrophotometric methods, peak shape serves as a critical performance indicator. Symmetrical, Gaussian peaks are not merely an aesthetic ideal but are fundamental for achieving accurate integration, reliable quantitation, and high resolution, directly impacting the correctness of method discrimination conclusions [81] [82]. Peak shape distortions introduce errors in peak area and height measurements, compromise resolution between closely eluting compounds, and can lead to misinterpretation of data, particularly when comparing the inherent performance of different analytical techniques [81] [83]. This application note provides a detailed protocol for diagnosing and resolving the most common peak shape anomalies—tailing, fronting, and broadening—within the context of advanced pharmaceutical analysis.
Understanding and Quantifying Peak Shape
Defining Ideal and Abnormal Peaks
A perfectly symmetrical peak is described as Gaussian. Deviations from this ideal manifest primarily as tailing, where the posterior half of the peak is broader, or fronting (also called leading), where the anterior half is broader [81] [84]. Broadening can occur symmetrically or asymmetrically, while splitting appears as a shoulder or twin peak on what should be a single component [81] [85].
Quantitative Measures of Peak Shape
Two primary metrics are used to quantify peak asymmetry, as illustrated in Table 1. Consistent tracking of these values is a cornerstone of robust system suitability tests [83].
Table 1: Quantitative Measures of Peak Asymmetry
| Measure | Calculation Formula | Measurement Height | Ideal Value | Typical Acceptance Limit |
|---|---|---|---|---|
| USP Tailing Factor (Tf) | Tf = (a + b) / 2a | 5% of peak height | 1.0 | Generally ≤ 2.0 [83] |
| Asymmetry Factor (As) | As = b / a | 10% of peak height | 1.0 | Generally ≤ 1.5 for many assays [86] |
(a = front half-width, b = back half-width)
For a more comprehensive analysis beyond single-value descriptors, advanced techniques like Total Peak Shape Analysis can be employed. This involves graphical tests, such as the derivative test, to detect and quantify concurrent fronting and tailing that might otherwise go unnoticed [82] [87].
Diagnosis and Resolution of Peak Abnormalities
The following troubleshooting workflow provides a systematic approach for diagnosing and correcting peak shape issues. This logical sequence guides the researcher from initial observation to a resolved method.
Peak Tailing
Peak tailing, characterized by an asymmetry factor (As) greater than 1.2, is the most common peak shape distortion [86].
Primary Causes and Protocols:
- Secondary Silanol Interactions: Basic analytes can strongly interact with acidic silanol groups on the silica surface.
- Protocol A (Lower pH): Adjust the mobile phase to a pH ≤ 3.0 to protonate silanol groups. Use a column stable at low pH (e.g., Agilent ZORBAX Stable Bond). Note: Standard silica columns can dissolve at pH < 3 [81] [86].
- Protocol B (Highly Deactivated Column): Employ an "end-capped" column (e.g., Agilent ZORBAX Eclipse Plus) where residual silanols are converted to less polar functional groups [81] [86].
- Protocol C (Buffering): Add a 5-10 mM buffer to the mobile phase to control pH and mask silanol interactions [81] [83].
- Column Overload: Occurs when the sample mass exceeds the column's capacity.
- Protocol D (Assess Load): Dilute the sample 10-fold and re-inject. If tailing is reduced, the column was overloaded.
- Protocol E (Reduce Load): Permanently resolve by reducing the injection volume, diluting the sample, or using a column with higher capacity (e.g., larger diameter, higher % carbon) [81] [86].
- Column Bed Deformation: Voids or channels in the packing bed disrupt flow paths.
- Protocol F (Diagnose): Substitute the column with a new one. If the problem is resolved, the original column is compromised.
- Protocol G (Rectify): Reverse the column and flush with a strong solvent (≥10 column volumes). If ineffective, replace the column. Use in-line filters and guard columns prophylactically [81] [86].
Peak Fronting
Peak fronting occurs when the front half of the peak is broader than the rear half.
Primary Causes and Protocols:
- Column Overload / Saturation: An overloaded mobile phase or stationary phase can cause molecules to elute faster.
- Sample Solvent Mismatch: The sample solvent is stronger than the initial mobile phase.
- Column Collapse / Deterioration: A sudden physical change in the column bed structure.
Peak Broadening and Splitting
Broadening reduces efficiency, while splitting can indicate multiple unresolved issues.
Primary Causes and Protocols:
- Extra-column Volume: Excessive dead volume in tubing connections between the injector, column, and detector.
- Protocol K (Inspect Connections): Ensure all tubing is cut squarely and inserted fully into fittings until it presses against the far side [88].
- Blocked Frit or Column Voids: Particulate matter blocking the inlet frit or voids in the packing material cause uneven flow paths.
- Co-elution: A single, asymmetric peak may actually be two or more poorly resolved compounds.
Table 2: Troubleshooting Guide for Poor Peak Shapes
| Symptom | Likely Cause | Recommended Experiments & Solutions |
|---|---|---|
| Tailing of one or a few peaks | Secondary silanol interactions (for basic compounds) | 1. Lower mobile phase pH to ~3.0 [86].2. Switch to a highly deactivated, end-capped column [81] [86].3. Increase buffer concentration (5-10 mM) [83]. |
| Tailing of all peaks | Column mass overload | 1. Dilute sample 10-fold and re-inject [86].2. Reduce injection volume or mass loaded [81]. |
| Column bed deformation (void) | 1. Replace with a new column to confirm [81].2. Reverse and flush the column with strong solvent [86]. | |
| Peak Fronting | Column overload / saturation | 1. Reduce injection volume or sample concentration [81] [84].2. Use a thicker film (GC) or larger ID column [84]. |
| Sample solvent mismatch | 1. Re-prepare sample in a solvent that matches the initial mobile phase strength [84] [88]. | |
| Column collapse | 1. Replace the column. Operate within the column's recommended pH and temperature limits [81] [83]. | |
| Peak Splitting | Blocked inlet frit or column void | 1. Replace the frit or the entire column [81] [85].2. Use an in-line filter and guard column. |
| Co-elution of compounds | 1. Inject a smaller volume to check for resolution [85].2. Adjust method parameters (temperature, gradient) to improve separation [85]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
The following reagents and materials are critical for executing the diagnostic and corrective protocols outlined in this document.
Table 3: Essential Research Reagents and Materials for Peak Shape Correction
| Item | Function / Application |
|---|---|
| End-capped C18 Column (e.g., ZORBAX Eclipse Plus) | Highly deactivated stationary phase to minimize secondary silanol interactions with basic analytes, reducing tailing [86]. |
| Low-pH Stable Column (e.g., ZORBAX Stable Bond) | For separations requiring operation at pH ≤ 3 to suppress silanol activity without damaging the silica backbone [86]. |
| Ammonium Formate/Acetate Buffers | To prepare buffered mobile phases (typically 5-10 mM) for controlling pH and masking silanol interactions [81] [83]. |
| Phosphoric Acid / Trifluoroacetic Acid (TFA) | For precise adjustment of mobile phase to low pH (e.g., pH 2.5-3.0) [86]. |
| In-line Filters (0.5 µm) & Guard Columns | Placed before the analytical column to trap particulates and protect the column from contaminants, preventing frit blockage and bed deformation [81] [86]. |
| UPLC/HPLC-grade Solvents (Water, Acetonitrile, Methanol) | For mobile phase and sample preparation to ensure purity, prevent contamination, and maintain system stability. |
| Polypropylene Syringes & Nylon Membranes (0.45 µm, 25 mm) | For sample filtration prior to injection, as referenced in IVRT method development [89]. |
Mastering the diagnosis and correction of poor peak shapes is non-negotiable for generating high-quality, reliable chromatographic data. The systematic protocols and detailed methodologies provided here empower researchers to not only troubleshoot effectively but also to develop more robust methods from the outset. In the critical comparison of UFLC against spectrophotometric techniques, where resolution, sensitivity, and quantitative accuracy are paramount, optimal peak shape is a fundamental determinant of valid and defensible scientific conclusions.
In the context of analytical method discrimination research, particularly when comparing ultra-fast liquid chromatography (UHPLC) to spectrophotometric techniques, understanding and controlling instrumental errors is paramount. For spectrophotometric methods, stray light and wavelength inaccuracy represent two critical sources of error that can compromise data integrity, especially when validating against more selective chromatographic methods [42] [71]. Stray light causes negative deviations from the Beer-Lambert law, particularly at high absorbance values, leading to inaccurate concentration measurements [90] [91]. Wavelength inaccuracy directly affects method specificity by shifting absorbance readings, which can alter calculated concentrations and invalidate method comparisons [92] [93]. This application note provides detailed protocols to identify, quantify, and mitigate these errors to ensure spectrophotometric data reliability in pharmaceutical analysis and development.
Theoretical Background
Stray Light: Origins and Impact
Stray light is defined as any light reaching the detector that lies outside the wavelength band width selected for analysis by the monochromator [90]. It arises from multiple sources including light scatter, diffraction by optical components, imperfections in optical surfaces, or even from the sample itself [91].
The primary effect of stray light is to reduce observed absorbance, leading to a negative deviation from Beer-Lambert law [90] [91]. This occurs because stray light constitutes an unabsorbed component of the total light reaching the detector, making samples appear more transparent than they truly are. The relative error becomes most significant at high absorbance values where the stray light component constitutes a larger fraction of the total transmitted light [90]. As noted in comparative studies, stray light can cause coefficients of variation in transmittance of up to 11% among different laboratories [92].
Wavelength Inaccuracy: Consequences for Specificity
Wavelength inaccuracy refers to the discrepancy between the wavelength selected on the instrument and the actual wavelength of light passing through the sample. This error affects both the sensitivity and the specificity of spectrophotometric methods [92] [93].
Inaccurate wavelength selection is particularly problematic when measuring at absorption maxima slopes, where small shifts can cause substantial changes in measured absorbance [92]. For pharmaceutical methods requiring discrimination between techniques, this error can directly impact the validity of comparative studies against UHPLC, which typically offers superior specificity through separation and mass spectrometric detection [42].
Experimental Protocols
Protocol 1: Stray Light Assessment and Control
Principle
Stray light is quantified using cut-off filters that absorb light completely at the wavelength of measurement but transmit at higher wavelengths. Any light detected below these cut-off wavelengths is by definition stray light [90].
Materials and Reagents
- High-quality spectrophotometer with deuterium and tungsten lamps
- Quartz cuvettes (path length 10 mm)
- Stray light calibration solutions:
- Sodium iodide (10 g/L) for 220 nm measurement
- Sodium nitrite (50 g/L) for 340 nm and 370 nm measurement
- Alternatively: Potassium chloride (12 g/L) for measurement at 198 nm (Pharmacopoeial procedure) [90]
Procedure
- Instrument Warm-up: Allow the spectrophotometer to warm up for at least 30 minutes to stabilize the light source and detector.
- Baseline Correction: Perform a baseline correction with a matched quartz cuvette containing high-purity water or the appropriate solvent.
- Solution Preparation: Precisely prepare fresh cut-off filter solutions using high-purity reagents and spectrophotometric-grade water.
- Measurement:
- Fill a clean quartz cuvette with the appropriate cut-off solution.
- Measure the transmittance at the specified wavelength (e.g., 220 nm for sodium iodide).
- Any detected transmittance greater than 0.1% indicates significant stray light [90].
- Acceptance Criteria: For a high-quality instrument, stray light should be <0.1% over the entire wavelength range [91].
Mitigation Strategies
- Regular Maintenance: Clean optical components (lenses, mirrors) regularly with lint-free cloths and appropriate solvents [93].
- Light Source Replacement: Replace deuterium and tungsten-halogen lamps according to manufacturer specifications or when performance degrades [93].
- Optical Alignment: Periodically verify and adjust instrument alignment to minimize diffraction effects [93].
- Environmental Control: Ensure the instrument is located in a low-light environment and the sample compartment is kept clean and free of reflective surfaces [91].
Protocol 2: Wavelength Accuracy Verification
Principle
Wavelength accuracy is verified using reference materials with sharp, well-characterized absorption or emission peaks at known wavelengths. Holmium oxide filters or solution provide multiple sharp peaks across UV and visible regions, while didymium filters offer broader bands suitable for initial checks [92] [93].
Materials and Reagents
- Holmium oxide filter or holmium oxide solution (4% in perchloric acid)
- Didymium filter
- Quartz cuvettes (path length 10 mm)
- Emission line sources (deuterium or hydrogen lamp) for instruments with this capability
Procedure
- Instrument Setup: Set the spectrophotometer to the slowest available scan speed and minimum slit width for maximum resolution.
- Holmium Oxide Verification:
- Place the holmium oxide filter in the light path or fill a cuvette with holmium oxide solution.
- Scan from 200 nm to 650 nm.
- Record the wavelengths of key absorption peaks (e.g., 241.1 nm, 287.1 nm, 361.5 nm, 536.2 nm).
- Compare measured peak wavelengths against certified values [92].
- Emission Line Method (if applicable):
- Replace the continuous light source with a deuterium or hydrogen emission lamp.
- Scan across known emission lines (deuterium: 656.1 nm, 486.0 nm).
- Record the detected wavelengths and compare to reference values [92].
- Acceptance Criteria: The measured peak wavelengths should typically be within ±0.5 nm of the certified values for UV-Vis spectrophotometers used in pharmaceutical analysis [93].
Error Correction
- Calibration Adjustment: If systematic error is detected, follow manufacturer procedures for wavelength calibration.
- Software Correction: Implement mathematical correction factors in data processing software if hardware calibration is not feasible.
- Preventive Maintenance: Establish regular verification schedules based on instrument usage and environmental conditions.
Table 1: Standard Reference Peaks for Wavelength Accuracy Verification
| Reference Material | Characteristic Peak Wavelengths (nm) | Tolerance | Application |
|---|---|---|---|
| Holmium Oxide Filter | 241.1, 287.1, 361.5, 536.2 | ±0.5 nm | Primary standard, full range |
| Holmium Oxide Solution | 241.1, 287.1, 361.5, 536.2 | ±1.0 nm | Alternative to filter |
| Didymium Filter | 528.7, 586.2 (broad peaks) | ±2.0 nm | Routine check, visible region |
| Deuterium Emission | 486.0, 656.1 | ±0.2 nm | Highest precision |
Research Reagent Solutions
Table 2: Essential Materials for Spectrophotometric Performance Verification
| Reagent/Standard | Function | Key Application | Specifications |
|---|---|---|---|
| Holmium Oxide Filter | Wavelength accuracy verification | Provides sharp, known absorption peaks across UV-Vis spectrum | NIST-traceable certified values |
| Potassium Chloride (12 g/L) | Stray light quantification | Measurement at 198 nm per Pharmacopoeial method | Absorbance ≥2.0 at 198 nm |
| Sodium Iodide (10 g/L) | Stray light assessment | UV region verification (220 nm) | Sharp cut-off below 260 nm |
| Sodium Nitrite (50 g/L) | Stray light assessment | Higher wavelength verification (340 nm, 370 nm) | Cut-off filter solution |
| Certified Quartz Cuvettes | Sample containment | Ensure path length accuracy and minimal light scatter | 10 mm path length, ±0.01 mm tolerance |
| Neutral Density Filters | Photometric linearity verification | Absorbance accuracy across working range | Certified absorbance values at specified wavelengths |
Method Discrimination Context: Spectrophotometry vs. UHPLC
In comparative studies between ultra-fast liquid chromatography and spectrophotometry, controlling spectrophotometric errors becomes crucial for valid method discrimination. UHPLC with mass spectrometric detection offers inherent advantages in specificity and sensitivity, capable of detecting compounds at ng/L levels with minimal interference [42]. Spectrophotometry, while simpler and more cost-effective, is more susceptible to matrix effects and spectral interferences [71].
Stray light and wavelength inaccuracy specifically impact the linear dynamic range and specificity of spectrophotometric methods, potentially leading to erroneous conclusions in method comparison studies. Proper control of these parameters ensures that observed differences truly reflect methodological capabilities rather than instrumental artifacts [42] [71].
For drug development applications, where UHPLC-MS/MS is increasingly the gold standard for pharmaceutical contaminant detection, well-characterized spectrophotometric methods remain valuable for rapid screening and analysis where extreme sensitivity is not required [42]. The protocols described herein enable researchers to maintain spectrophotometric data quality sufficient for meaningful comparison with chromatographic techniques.
Effective mitigation of stray light and wavelength inaccuracy is fundamental to generating reliable spectrophotometric data, particularly in studies comparing analytical techniques. The protocols outlined provide researchers with standardized approaches to quantify and control these critical parameters. By implementing these verification procedures regularly and maintaining meticulous instrument records, laboratories can ensure the integrity of their spectrophotometric methods and enable meaningful discrimination between analytical techniques in pharmaceutical development and quality control environments.
Combating Matrix Effects and Ionization Suppression in LC-MS
Matrix effects pose a significant challenge in liquid chromatography-mass spectrometry (LC-MS) bioanalysis, particularly in pharmaceutical development where ultra-fast liquid chromatography methods are increasingly employed. These effects manifest as ion suppression or enhancement when co-eluting matrix components interfere with the ionization efficiency of target analytes in the mass spectrometer source [94]. This phenomenon represents a critical methodological consideration when comparing LC-MS to traditional spectrophotometric techniques, as matrix effects can severely impact the accuracy, precision, and sensitivity of quantitative results [95] [96].
The mechanisms underlying matrix effects vary based on ionization technique. In electrospray ionization (ESI), which is particularly susceptible, proposed mechanisms include competition for available charge at the droplet surface, changes in solution viscosity affecting droplet formation, and neutralization of ions in the gas phase [97] [94]. Atmospheric pressure chemical ionization (APCI) generally demonstrates less susceptibility to matrix effects, as the ionization process occurs in the gas phase rather than in condensed droplets [97] [94]. Biological matrices introduce numerous potential interferents, including phospholipids, salts, carbohydrates, lipids, peptides, and metabolites, along with exogenous compounds from sample handling such as polymers from plastic materials and anticoagulants [97] [98].
Detection and Assessment Methods
Qualitative Assessment: Post-Column Infusion
The post-column infusion method provides a qualitative assessment of matrix effects throughout the chromatographic run [98] [94].
Experimental Protocol:
- Connect a syringe pump containing a solution of the analyte of interest to the system via a tee-fitting between the chromatographic column outlet and the MS inlet
- Infuse the analyte at a constant rate to establish a stable baseline signal
- Inject a prepared blank matrix extract onto the LC column
- Monitor the analyte signal throughout the chromatographic run
Interpretation: Regions where the constant signal decreases indicate ion suppression, while signal increases indicate ion enhancement [94]. This method effectively maps problematic retention time windows but does not provide quantitative assessment of matrix effects [95].
Quantitative Assessment: Post-Extraction Spiking
The post-extraction spiking approach, introduced by Matuszewski et al., provides quantitative assessment of matrix effects and is considered the "golden standard" in regulated bioanalysis [97] [98].
Experimental Protocol:
- Prepare at least six lots of blank matrix from different sources
- Extract each matrix lot using the intended sample preparation method
- Spike the analyte at known concentrations (typically low and high QC levels) into the post-extraction blanks
- Prepare equivalent standards in neat solution
- Analyze all samples and calculate the matrix factor (MF) using the formula: MF = Peak area of analyte in post-extracted spiked matrix / Peak area of analyte in neat solution [98]
Interpretation: MF values <1 indicate ion suppression, >1 indicate ion enhancement, and ≈1 indicate no significant matrix effects [97]. The internal standard-normalized MF (MFanalyte/MFIS) should be close to 1.0 for proper compensation, with values ideally between 0.75-1.25 for robust methods [98].
Table 1: Comparison of Matrix Effect Assessment Methods
| Method | Type of Data | Key Advantages | Key Limitations | Regulatory Status |
|---|---|---|---|---|
| Post-Column Infusion | Qualitative | Identifies suppression/enhancement regions; Guides method development | Does not provide quantitative measurement; Requires additional hardware | Recommended during method development |
| Post-Extraction Spiking | Quantitative | Provides numerical matrix factor; Assesses lot-to-lot variability | Requires multiple matrix lots; Does not identify specific interferents | Golden standard in regulated bioanalysis (ICH M10) |
| Pre-Extraction Spiking | Qualitative | Demonstrates method consistency; Uses standard validation samples | Does not quantify matrix effect magnitude | Required per ICH M10 guideline |
Strategic Approaches for Mitigation
Sample Preparation Optimization
Effective sample preparation serves as the first line of defense against matrix effects by removing potential interferents prior to analysis [97] [95].
Supported Liquid Extraction (SLE) has demonstrated superior performance for vitamin E analysis in plasma, providing cleaner extracts compared to protein precipitation, solid-phase extraction, and liquid-liquid extraction [97]. The selectivity of the sample preparation method should be matched to the complexity of the matrix, with biological samples typically requiring more extensive clean-up.
Sample dilution represents a straightforward approach when assay sensitivity permits, effectively reducing the concentration of interferents [95]. For studies anticipating significant matrix effects (e.g., from intravenous administration with vehicles containing PEG-400 or Tween-80), pre-dilution of study samples is recommended, particularly for early time points [98].
Chromatographic Method Development
Chromatographic separation directly impacts matrix effects by determining the extent of co-elution between analytes and matrix components [95]. Ultra-fast liquid chromatography methods require particular attention to this aspect, as compressed separation windows may increase co-elution risks.
Strategy for chromatographic optimization:
- Extend separation when possible to resolve analytes from interferents
- Alter selectivity through changes in stationary phase chemistry
- Utilize supercritical fluid chromatography (SFC) as an alternative separation mechanism with different selectivity that can separate compounds causing matrix effects in LC [97]
- Adjust retention to elute analytes in regions with minimal suppression, as identified by post-column infusion
Mobile phase additives should be selected carefully, as some additives can act as ion-pairing reagents or directly suppress ionization [95].
Ionization Technique Selection
The choice of ionization technique significantly influences susceptibility to matrix effects [94].
ESI to APCI switching represents a valuable strategy when matrix effects persist despite sample preparation and chromatographic optimization. APCI typically demonstrates reduced matrix effects because ionization occurs in the gas phase rather than in liquid droplets, minimizing competition effects [94]. However, APCI has limitations for non-volatile or thermally labile compounds [98].
Atmospheric pressure photoionization (APPI) provides an alternative for less polar compounds that may not ionize efficiently by ESI or APCI [74]. The selection of ionization polarity (positive vs. negative) also affects matrix effect susceptibility, with fewer compounds typically responding in negative mode [94].
Calibration Strategies for Compensation
When matrix effects cannot be sufficiently eliminated, calibration strategies provide compensation during quantitative analysis [95].
Stable isotope-labeled internal standards (SIL-IS) represent the optimal approach for compensation, as they possess nearly identical chemical properties to the analytes and co-elute chromatographically, experiencing virtually the same matrix effects [97] [95] [98]. The IS-normalized matrix factor should be close to 1.0, demonstrating effective compensation [98].
Alternative compensation methods include:
- Matrix-matched calibration: Preparing standards in the same matrix as samples [97]
- Standard addition: Adding known amounts of analyte to sample aliquots [95]
- Structural analogue internal standards: Using chemically similar compounds when SIL-IS are unavailable [95]
Table 2: Matrix Effect Mitigation Strategies and Applications
| Strategy | Mechanism of Action | Best Applications | Practical Considerations |
|---|---|---|---|
| Supported Liquid Extraction | Selective removal of phospholipids and endogenous interferents | Complex biological matrices (plasma, serum) | Superior recovery and cleaner extracts vs. PPT and LLE |
| UPLC/UHPLC Separation | Improved chromatographic resolution to separate analytes from interferents | Methods requiring high sensitivity and specificity | Enhanced peak capacity; compatible with fast analysis |
| SFC-MS | Alternative separation mechanism with different selectivity | Problematic analytes with significant LC-MS matrix effects | Different elution profile for matrix components |
| SIL-Internal Standards | Co-elution with analytes for identical matrix effects | Quantitative bioanalysis where available | Optimal compensation but expensive; not always available |
| ESI to APCI Switching | Gas-phase ionization reduces competition effects | Compounds amenable to APCI | Not suitable for non-volatile or thermally labile compounds |
Experimental Protocols
Comprehensive Matrix Effect Assessment Protocol
This integrated protocol combines qualitative and quantitative assessment approaches for thorough matrix effect evaluation during method development.
Materials and Reagents:
- Blank matrix from at least six individual sources
- Analytes of interest and appropriate internal standards
- All HPLC/MS-grade solvents and reagents
- Syringe pump for post-column infusion
Procedure:
- Perform post-column infusion to identify regions of ion suppression/enhancement
- Optimize chromatographic conditions to elute analytes in minimal suppression regions
- Prepare post-extraction spiked samples using six matrix lots at low and high QC concentrations
- Analyze alongside neat standards to calculate absolute matrix factors
- Calculate IS-normalized MF to assess compensation efficiency
- Validate method accuracy using pre-extraction spiked QCs in all matrix lots
Acceptance Criteria: IS-normalized MF should be 0.75-1.25 with CV ≤15%; pre-extraction spiked QC accuracy should be within ±15% of nominal values [98].
Sample Preparation Protocol Using Supported Liquid Extraction
Reagents and Materials:
- Supported liquid extraction plates
- Appropriate organic solvents (MTBE, hexane, ethyl acetate)
- Evaporation system (nitrogen or air)
- Reconstitution solution
Procedure:
- Aliquot 100-200 μL of sample (plasma, serum)
- Pre-treat with appropriate solution (e.g., formic acid solution)
- Load onto pre-conditioned SLE plate
- Wait 5-10 minutes for complete absorption
- Elute with organic solvent (e.g., 2 × 1 mL methyl tert-butyl ether)
- Evaporate eluent to dryness under nitrogen stream
- Reconstitute in mobile phase compatible solution
- Analyze by LC-MS
This SLE protocol has demonstrated superior performance for challenging analyses such as vitamin E forms in plasma, providing cleaner extracts and reduced matrix effects compared to alternative techniques [97].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Matrix Effect Investigation
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Stable Isotope-Labeled Internal Standards | Compensation for matrix effects during quantification | Optimal when co-elutes with analyte; should be added before sample preparation |
| Supported Liquid Extraction Plates | Sample clean-up to remove phospholipids and interferents | Superior to protein precipitation for reducing matrix effects |
| Polymer-Free Collection Tubes | Prevention of exogenous contaminant introduction | Avoids polyethylene glycol contamination that causes suppression |
| LC-MS Grade Solvents | Minimize background interference and signal noise | Reduces chemical noise that exacerbates matrix effects |
| Phospholipid Removal Plates | Selective removal of phospholipids from biological samples | Targets primary cause of matrix effects in plasma/serum |
| Matrix Matched Calibration Standards | Compensation for residual matrix effects | Requires significant amounts of blank matrix; matching critical |
Operational Recommendations and Monitoring
Method Development Workflow
The following workflow diagram illustrates the systematic approach to addressing matrix effects during method development:
Incurred Sample Analysis Monitoring
During study sample analysis, continuous monitoring of internal standard responses is critical for detecting subject-specific matrix effects not observed in validation [98]. Abnormal IS responses may indicate unexpected matrix effects from dosing vehicles, metabolites, or co-medications.
Protocol for investigation:
- Flag samples with IS responses ±3× from mean
- Reanalyze with greater dilution (if sensitivity permits)
- Compare original and reanalyzed concentrations
- Accept if within ±20% difference, indicating no significant impact on quantification
For studies with anticipated matrix effects (e.g., from intravenous formulations with solubilizing agents), pre-dilution protocols should be implemented for early time points [98].
Matrix effects remain a significant challenge in LC-MS bioanalysis, particularly with the implementation of ultra-fast chromatographic methods in pharmaceutical development. A systematic approach incorporating rigorous assessment during method development, strategic implementation of mitigation techniques, and vigilant monitoring during sample analysis represents the most effective strategy for combating ionization suppression and enhancement. The comprehensive protocols and strategies outlined herein provide a framework for developing robust LC-MS methods that deliver accurate and reliable quantitative results, underscoring the critical advantages of sophisticated LC-MS approaches over traditional spectrophotometric methods for complex matrix analysis.
Optimizing Mobile Phase and Column Temperature for Better Resolution
In the context of ultra-fast liquid chromatography (UFLC) versus spectrophotometric method discrimination research, achieving superior resolution is paramount. Resolution, the ability to distinguish between closely eluting peaks, is a critical performance metric that directly impacts the accuracy and reliability of analytical results, particularly in complex fields like drug development [34]. UFLC leverages high pressures and sub-2 µm particle columns to dramatically reduce analysis times compared to traditional HPLC, but this speed must not come at the cost of chromatographic resolution [20] [99]. Unlike spectrophotometric methods which may struggle with overlapping spectra in complex mixtures, a well-optimized UFLC method can separate and quantify individual components with high specificity.
Two of the most powerful and controllable parameters for enhancing resolution in UFLC are the mobile phase composition and the column temperature [13]. Systematic optimization of these factors allows researchers to fine-tune the interactions between analytes, the mobile phase, and the stationary phase, thereby achieving the desired separation efficiency. This document provides detailed application notes and protocols for optimizing these parameters to maximize resolution within the fast analysis frameworks required by modern pharmaceutical and bioanalytical research.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and reagents essential for executing the optimization protocols described in this document.
Table 1: Key Research Reagent Solutions for UFLC Optimization
| Item | Function/Description | Application Note |
|---|---|---|
| Ammonium Formate/Acetate | Mobile phase additive to improve ionization efficiency and control pH [100]. | Essential for LC-MS compatibility; reduces ion suppression and enhances signal stability. |
| Formic Acid/Acetic Acid | Mobile phase modifier to provide acidic pH and influence analyte ionization [100]. | Critical for controlling retention and peak shape of acidic and basic compounds. |
| High-Purity Water & Organic Solvents (ACN, MeOH) | The primary constituents of the mobile phase [1]. | Must be LC-MS grade and filtered to 0.2 µm to prevent system damage and background noise. |
| Sub-2 µm Particle UHPLC Columns | Stationary phase for achieving high-efficiency separations [99]. | Core component for UFLC; provides high peak capacity and fast separations under high pressure. |
| Inert (Biocompatible) UHPLC Columns | Columns with passivated hardware to minimize metal-analyte interactions [72]. | Crucial for analyzing metal-sensitive compounds like phosphorylated molecules, improving peak shape and recovery. |
| Trap Columns | Used for online sample clean-up and concentration [101]. | Removes salts and impurities from biological samples, mitigating matrix effects and protecting the analytical column. |
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Internal standards for quantitative mass spectrometry [1]. | Corrects for analyte loss during sample preparation and compensates for matrix-induced ion suppression/enhancement. |
| 0.2 µm Membrane Filters | For filtering mobile phases and samples [99]. | Mandatory for UHPLC to prevent clogging of columns and system components due to small particle sizes. |
Mobile Phase Optimization for Enhanced Selectivity
The mobile phase is not merely a carrier in chromatography; it is an active participant in the separation process. Its composition directly influences selectivity (α), which is a primary factor in achieving resolution. Optimization involves selecting the correct buffers, pH, and organic modifiers to fine-tune the interactions of analytes with the stationary phase.
Experimental Protocol: Systematic Mobile Phase Scouting
Objective: To identify the optimal mobile phase composition for resolving a complex mixture of polar metabolites and lipids. Materials: UHPLC system capable of withstanding pressures up to 1000 bar; HILIC and reversed-phase (e.g., C18) columns (sub-2 µm); your mixture of analytes; ammonium formate, ammonium acetate, formic acid, acetic acid, HPLC-grade water, acetonitrile, and methanol. Procedure:
- Sample Preparation: Dissolve the target analytes in a suitable solvent, typically the starting mobile phase composition. Centrifuge or filter through a 0.2 µm membrane to remove particulates [99].
- System Setup: Equip the UHPLC system with the appropriate column (HILIC for polar metabolites, C18 for lipids and less polar compounds). Ensure the system is purged and equilibrated.
- Scouting Gradient Run: Begin with a generic, broad gradient (e.g., 5% to 95% organic solvent over 10 minutes) to determine the approximate retention window of your analytes.
- Modifier Screening: Perform a series of isocratic or shallow gradient runs, testing different mobile phase modifiers as detailed in Table 2. For instance, compare the chromatographic performance of 10 mM ammonium formate with 0.125% formic acid against 10 mM ammonium acetate with 0.1% acetic acid [100].
- Data Collection and Analysis: Monitor key performance indicators including peak shape (asymmetry factor), signal intensity (for MS detection), retention time stability, and most critically, the resolution (Rs) between critical analyte pairs.
Table 2: Optimized Mobile Phase Modifiers for Different Applications [100]
| Analyte Class | Chromatography Mode | Recommended Mobile Phase Modifiers | Key Performance Benefits |
|---|---|---|---|
| Amino Acids, Sugars, Nucleotides | HILIC | 10 mM ammonium formate / 0.125% formic acid | Best overall performance for signal intensity |
| Organic Acids | Reversed-Phase (RPLC) | 0.1% formic acid | Outperforms other modifiers for this class |
| Lipids (ESI+ mode) | Reversed-Phase (RPLC) | 10 mM ammonium formate OR 10 mM ammonium formate / 0.1% formic acid | High signal intensity and robust retention times |
| Lipids (ESI- mode) | Reversed-Phase (RPLC) | 10 mM ammonium acetate / 0.1% acetic acid | Good compromise between signal intensity and retention time stability |
Logical Workflow for Mobile Phase Selection
The following diagram illustrates the decision-making process for selecting and optimizing the mobile phase, integrating the recommendations from the experimental data.
Column Temperature as a Tool for Efficiency and Resolution
Column temperature is a highly versatile yet often underutilized parameter in UFLC method development. Increasing the column temperature reduces mobile phase viscosity, which in turn lowers backpressure and accelerates mass transfer of analytes between the mobile and stationary phases [13]. This results in a "flatter" van Deemter curve, allowing the use of higher flow rates without a significant loss of efficiency, thereby enabling faster analyses. Furthermore, temperature can selectively alter the equilibrium constant of analytes, affecting retention and selectivity to improve resolution.
Experimental Protocol: Optimizing Temperature for Speed and Resolution
Objective: To determine the optimal column temperature for resolving a critical pair of compounds while minimizing analysis time. Materials: UHPLC system with a thermostatted column oven (forced-air is preferred); a suitable UHPLC column (sub-2 µm); your analyte mixture. Procedure:
- Initial Conditions: Set a fixed, moderate flow rate and mobile phase composition based on prior scouting.
- Temperature Gradient: Perform a series of injections at different column temperatures (e.g., 30°C, 40°C, 50°C, 60°C, 70°C). Ensure the system, including the mobile phase pre-heater, is fully equilibrated at each temperature.
- Data Collection: Record the retention times, peak widths, and resolution for the critical pair of analytes. Also, note the system backpressure at each temperature.
- Van Deemter Analysis (Optional but informative): For a more fundamental understanding, plot the height equivalent to a theoretical plate (HETP) against the linear velocity (flow rate) at two different temperatures (e.g., 30°C and 60°C). This will visually demonstrate how elevated temperature maintains efficiency at higher flow rates [13].
- Analysis: Identify the temperature that provides the best compromise of analysis speed (retention time), resolution, and backpressure. Be mindful of the thermal stability of both your analytes and the stationary phase.
Table 3: Quantitative Effects of Elevated Column Temperature [13]
| Parameter | Effect of Increasing Temperature | Impact on UFLC Performance |
|---|---|---|
| Mobile Phase Viscosity | Decreases | Allows for higher flow rates, reducing analysis time and lowering system backpressure. |
| Mass Transfer (C-term) | Accelerates | Reduces peak broadening, leading to higher column efficiency (more plates). |
| Analyte Retention (k') | Generally decreases | Can be used to shorten run times and modify selectivity for better resolution. |
| Backpressure | Decreases significantly (e.g., ~40% lower at 80°C vs 40°C) | Enables use of longer columns or higher flow rates within pressure limits. |
Integrated Workflow for Simultaneous Optimization
The most effective approach to method development involves understanding the interaction between mobile phase composition and column temperature. The following integrated protocol provides a systematic pathway.
Comprehensive Optimization Protocol
Objective: To establish a robust, high-resolution UFLC method by concurrently optimizing mobile phase and temperature. Materials: As in previous protocols. Procedure:
- Scouting and Screening: Follow the Mobile Phase Scouting Protocol (Section 3.1) to identify 2-3 promising mobile phase compositions.
- Temperature Profiling: For each promising mobile phase from Step 1, conduct the Temperature Optimization Protocol (Section 4.1) at three key temperatures (e.g., low, medium, high).
- Data Analysis and Selection: For each combination, calculate the resolution of all critical peak pairs and the total run time. Use this data to construct a simple response surface model to identify the "sweet spot."
- Final Method Validation: The optimal conditions should be validated for precision, accuracy, and robustness according to standard laboratory guidelines, paying particular attention to retention time stability and signal reproducibility [100].
Within the comparative framework of UFLC and spectrophotometric research, the ability to deliberately optimize chromatographic parameters is what grants UFLC its superior discriminatory power. While spectrophotometry may offer simplicity, it often falls short in deconvoluting complex samples. As detailed in these application notes, the strategic manipulation of mobile phase composition and column temperature provides researchers with a powerful and predictable means to maximize resolution without sacrificing the speed that defines ultra-fast chromatography. By adhering to the structured protocols and utilizing the essential tools outlined herein, scientists can develop robust, high-performance methods that meet the stringent demands of modern drug development and complex bioanalysis.
Preventing Sample Degradation and Photodegradation
Sample degradation, particularly photodegradation, presents a significant challenge in pharmaceutical analysis, directly impacting the accuracy, reliability, and reproducibility of analytical results. Within drug development, maintaining sample integrity from storage through analysis is paramount for correct quantification of active pharmaceutical ingredients (APIs) and assessment of stability. This document details practical protocols and strategies to mitigate these risks, framed within research comparing Ultra-Fast Liquid Chromatography (UFLC) and spectrophotometric methods. Understanding and controlling degradation pathways is essential for validating these analytical techniques, as degradation products can cause significant interference, especially in less selective spectrophotometric assays [102] [22].
Understanding Degradation Pathways
Degradation is a process where an API undergoes a chemical change under the influence of environmental factors such as light, heat, humidity, or oxygen. Photodegradation, a subset of this process, is a chemical change initiated by the absorption of light energy [103].
Key Photodegradation Mechanisms
There are two primary classes of photochemical reactions:
- Direct Photochemical Reactions: The analyte directly absorbs light, leading to reactions such as ionization, isomerization, or bond cleavage. The molecule's structure and the energy of the absorbed photon determine the specific reaction [103].
- Photosensitized Reactions: A photosensitizer (a chromophore present in the sample or matrix) absorbs light and transfers energy to the analyte, indirectly causing its degradation. This often occurs via photo-oxidation mechanisms [103].
- Type-I Mechanism: Involves hydrogen abstraction or electron transfer between the excited photosensitizer and the substrate, generating free radicals. This is more common under lower oxygen conditions [103].
- Type-II Mechanism: The excited photosensitizer energizes molecular oxygen to highly reactive singlet oxygen, which then attacks the analyte. This is the dominant pathway in aerobic environments [103].
Other common reactions include photo-isomerization, where a molecule changes its isomeric form (e.g., trans to cis), and photolytic auto-oxidation, prevalent in lipids and carotenoids [103].
Experimental Protocols for Degradation Studies
Forced degradation studies are critical for understanding the stability of an analyte and for validating the stability-indicating properties of an analytical method.
Protocol 1: Forced Photodegradation Study
This protocol outlines a procedure to assess the photosensitivity of an API, based on studies of compounds like candesartan cilexetil and steroid hormones [102] [104].
- Objective: To determine the rate and extent of degradation of an API under controlled light exposure and to identify resulting degradation products.
Materials:
- API (pure substance or extracted from formulation)
- Appropriate solvent (e.g., methanol, water, or mixture)
- Quartz or clear glass petri dishes/vials (for UV transparency)
- Controlled light source (e.g., UV lamp, sunlight simulator, or direct sunlight)
- Aluminum foil (for dark control)
- UFLC-DAD/MS system or spectrophotometer
Procedure:
- Sample Preparation: Prepare a solution of the API at a known concentration (e.g., 100 µg/mL) in a suitable solvent.
- Exposure: Divide the solution into multiple aliquots. Place these in petri dishes, ensuring a large surface-to-volume ratio to enhance light penetration [104].
- Light Stress: Expose the samples to the light source. For a systematic study, use a range of light intensities and exposure durations (e.g., from 6 hours to several days), withdrawing samples at regular intervals [102].
- Dark Control: Protect an identical sample aliquot from light using aluminum foil and store it under otherwise identical conditions.
- Analysis: Withdraw samples at each time point and dilute as necessary. Analyze both stressed and control samples using UFLC-DAD/MS and UV spectrophotometry.
- Data Analysis: Calculate the percentage of API remaining at each interval. UFLC-DAD/MS allows for the identification and tracking of specific degradation products via their unique retention times and mass spectra [102] [104].
Protocol 2: Comparative Analysis of UFLC vs. Spectrophotometry for Degradation Monitoring
This protocol evaluates the ability of UFLC and spectrophotometric methods to discriminate between the intact API and its degradation products.
- Objective: To compare the discrimination power of UFLC and UV spectrophotometry in quantifying an API in the presence of its degradation products.
Materials:
- Degraded API samples (from Protocol 1)
- Standard solutions of pure API
- UFLC-DAD/MS system
- UV-Vis spectrophotometer
Procedure:
- Calibration: Construct calibration curves for the pure API using both UFLC (peak area) and UV spectrophotometry (absorbance).
- Analysis of Degraded Samples: Analyze the degraded samples from Protocol 1 using both techniques.
- Specificity/Selectivity Assessment:
- UFLC-DAD/MS: Examine chromatograms for the resolution of the API peak from degradation product peaks. Use DAD UV spectra and MS data to confirm the purity and identity of each peak [22] [105].
- UV Spectrophotometry: Measure the absorbance of the degraded sample at the λmax of the API. The presence of overlapping absorbances from degradation products will lead to an overestimation of the API concentration [22] [64].
- Data Comparison: Compare the calculated concentration of the API in the degraded samples obtained from both methods. The UFLC method, with its separation power, should provide a more accurate quantification of the remaining intact API, while the spectrophotometric method will often report a higher, inaccurate value due to combined absorbances.
Key Data and Comparative Analysis
Quantitative Degradation Data
The following table summarizes exemplary degradation data for an API under various stress conditions, illustrating the extent of degradation measurable by analytical methods.
Table 1: Exemplary Degradation Profile of an API under Stress Conditions (based on candesartan cilexetil data) [102]
| Stress Condition | Details | % API Remaining | Major Degradation Products | Analysis Method |
|---|---|---|---|---|
| Acidic Hydrolysis | 0.1 N HCl, 60°C, 5 hours | ~20% | Acidic degradants | UV at λmax |
| Alkaline Hydrolysis | 0.1 N NaOH, 60°C, 5 hours | ~85% | Basic degradants | UV at λmax |
| Oxidative Degradation | 3% H₂O₂, dark, 12 hours | ~40% | Hydroperoxides | UV at λmax |
| Thermal Degradation | Dry heat, 60°C, 3 days | ~70% | Thermal degradants | UV at λmax |
| Photolytic Degradation | Direct sunlight, 3 days | ~90% | Isomers, photo-oxidants | UV at λmax |
| UV Light Degradation | UV lamp, 24 hours | ~95% | Photo-oxidants | UV at λmax |
Method Comparison for Degradation Analysis
The choice of analytical technique critically affects the interpretation of stability data.
Table 2: Comparison of UFLC and Spectrophotometry for Analyzing Degraded Samples [22] [64] [106]
| Parameter | Ultra-Fast Liquid Chromatography (UFLC) | UV-Vis Spectrophotometry |
|---|---|---|
| Selectivity | High (separates API from degradants) [22] | Low (measures total absorbance) [22] [64] |
| Specificity | High (DAD and MS detect co-eluting peaks) [22] [105] | Low (cannot distinguish between analytes) [64] |
| Sensitivity | High (detection limits in ng/mL) [22] [106] | Moderate (µg/mL range) [64] |
| Quantification Accuracy | Accurate for individual components in a mixture [22] | Inaccurate in mixtures without separation [22] |
| Degradation Product Identification | Yes (via retention time, DAD spectrum, MS) [102] [105] | No (only indicates overall change) [64] |
| Cost & Operational Complexity | High (equipment, solvents, trained personnel) [22] [106] | Low (simple, cost-effective, minimal training) [22] [64] |
| Greenness (AGREE metric) | Lower (higher solvent consumption) [22] | Higher (lower solvent consumption) [22] |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for Degradation Studies
| Item | Function & Application |
|---|---|
| Methanol:Water (9:1) | A common solvent system for UV analysis and sample preparation, providing good solubility for many APIs [102]. |
| Quartz Cuvettes/Cells | Essential for photodegradation studies in UV regions, as they are transparent to UV light, unlike many plastics [102]. |
| 0.1 N HCl & 0.1 N NaOH | Standard reagents for conducting acid and base hydrolysis forced degradation studies [102]. |
| 3% Hydrogen Peroxide (H₂O₂) | A standard oxidizing reagent used to simulate oxidative degradation pathways [102]. |
| Photosensitizers (e.g., Porphyrins) | Used in controlled studies to understand and amplify Type-I/II photo-oxidation degradation mechanisms [104]. |
| Complexing Agents (e.g., FeCl₃) | Used in spectrophotometric assays to form colored complexes with non-chromophoric analytes or degradation products for detection [64]. |
| Diazotization Reagents (NaNO₂/HCl) | Used to derivatize primary aromatic amines (a common functional group in APIs and degradants) for sensitive spectrophotometric detection [64]. |
Workflow and Pathway Visualizations
Photodegradation Experimental Workflow
Photodegradation Reaction Pathways
Preventing and accurately monitoring sample degradation is a cornerstone of robust pharmaceutical analysis. This document provides application notes and protocols demonstrating that while UFLC offers superior discrimination by separating and individually quantifying APIs and their degradation products, UV spectrophotometry serves as a rapid, cost-effective initial screening tool. The implementation of controlled forced degradation studies, as outlined, is vital for validating any analytical method. The choice between advanced techniques like UFLC and simpler spectrophotometric methods should be guided by the required level of specificity, available resources, and the criticality of the data, ensuring the integrity of results throughout the drug development pipeline.
Baseline Noise and Retention Time Shift Solutions
In the pursuit of high-throughput and high-resolution analytical data within pharmaceutical research and development, Ultra-Fast Liquid Chromatography (UFLC), particularly Ultra-High-Performance Liquid Chromatography (UHPLC), has become a cornerstone technique. Its advantages over traditional spectrophotometric methods are profound, offering superior selectivity, sensitivity, and the ability to analyze complex mixtures. However, the enhanced performance of UHPLC brings with it a heightened sensitivity to operational parameters, making it susceptible to issues such as baseline noise and retention time shifts. These anomalies can compromise data integrity, leading to inaccurate quantification and misinterpretation of results. This application note provides a detailed diagnostic and procedural guide for scientists to identify, troubleshoot, and resolve these critical issues, thereby ensuring the reliability of chromatographic data in fast-paced drug development environments.
Understanding and Diagnosing Chromatographic Issues
Baseline Noise
Baseline noise refers to the short-term, random fluctuations in the detector signal when only the mobile phase is flowing. It is a critical parameter as it directly impacts the Limit of Detection (LOD) and Limit of Quantification (LOQ). The signal-to-noise ratio (S/N) is a key metric, with an S/N of 3:1 typically defining the LOD and 10:1 the LOQ [107] [108]. Unusually high noise can obscure low-intensity peaks, such as impurities or degradants, rendering them undetectable and compromising method sensitivity.
Retention Time Shifts
Retention time (RT) stability is fundamental for peak identification and accurate integration. Shifts in RT can lead to misidentification of analytes or failure to detect them entirely. In UHPLC systems, which operate with smaller particle sizes and faster flow rates, the tolerances for such shifts are reduced, making the system more susceptible to minor disturbances [109].
The following tables consolidate key quantitative information and specifications relevant to diagnosing and resolving baseline noise and retention time issues.
Table 1: Signal-to-Noise Ratio Specifications for Detection and Quantification
| Parameter | S/N Ratio | Description | Reference Standard |
|---|---|---|---|
| Limit of Detection (LOD) | 3:1 | The minimum concentration at which an analyte can be reliably detected. | ICH Q2(R2) [107] |
| Limit of Quantification (LOQ) | 10:1 | The minimum concentration at which an analyte can be reliably quantified. | ICH Q2(R2) [107] [108] |
| Recommended Practical LOQ | 10:1 to 20:1 | A higher, more robust S/N ratio recommended for real-world sample analysis. | Industry Best Practice [107] |
Table 2: Common Causes and Solutions for Baseline Noise and Retention Time Shifts
| Symptom | Potential Cause | Recommended Solution | Experimental Verification |
|---|---|---|---|
| High-frequency baseline noise | Mobile phase not fully degassed [110] [108]; Dirty flow cell [108] | Degas mobile phase thoroughly (inline degasser, helium sparging) [111] [108]; Clean or replace flow cell windows [108] | Run a blank gradient; Measure noise with HPLC-grade water [112] [108] |
| Retention time shifts, stable pressure | Leaking needle seal [113] | Replace the needle seal; Purge fluidics; Perform dynamic leak test on injector pod [113] | Monitor RT consistency across a sequence; Perform system leak tests |
| RT shifts for particular analytes (e.g., amines) | Unstable pH due to residual additives [114] | Modify method to stabilize pH by adding a mobile phase modifier (e.g., formic acid, buffer) to all mobile phases [114] | Compare RT stability before and after adding a consistent modifier to all lines |
| Intermittent RT shifts (early eluting peaks) | Column degradation [115] | Replace the chromatographic column [115] | Evaluate system suitability standards; Check for peak splitting or loss of efficiency |
Experimental Protocols for Troubleshooting
Protocol 1: Systematic Diagnosis of Baseline Noise
This protocol provides a step-by-step methodology to identify and eliminate sources of excessive baseline noise.
4.1.1 Materials and Equipment
- HPLC/UHPLC system with UV or DAD detector
- Fresh, high-quality, and degassed mobile phases
- Isocratic method matching the initial conditions of your gradient method
- Post-detector pressure restrictor (if applicable)
4.1.2 Procedure
- Disconnect the Column: Replace the column with a zero-dead-volume union connector.
- Establish Baseline Conditions: Set the detector to the problematic wavelength (e.g., 214 nm for TFA). Run an isocratic method with a high proportion of aqueous phase (e.g., 95% Water, 5% Acetonitrile) at the standard flow rate.
- Observe and Document Baseline: Allow the system to stabilize for 15-20 minutes. Record the baseline for 10 minutes and calculate the peak-to-peak noise.
- Evaluate Detector Components:
- Lamp Age: Check the detector's lamp usage hours. Compare the current baseline noise to the instrument's specifications or historical data from a new lamp.
- Slit Width: If using a DAD, increase the slit width (e.g., from 1 nm to 4 nm). This increases light throughput, which can reduce noise but slightly decrease spectral resolution [108].
- Data Acquisition Rate: Increase the acquisition rate (e.g., to 20 Hz) to provide more data points for noise averaging, ensuring you maintain at least 20 points per peak [108].
- Assess Mixing Efficiency: For gradient methods, the noise may originate from poor mixing. Install a static mixer between the pump and the injector. Repeat the baseline observation and note any change in noise characteristics [111] [108].
- Reconnect Column: Once noise is minimized at the detector level, reconnect the column and repeat a blank injection to see if the column or sample contributes to the noise.
Protocol 2: Investigation and Resolution of Retention Time Shifts
This protocol is designed to diagnose the root cause of unstable retention times.
4.2.1 Materials and Equipment
- HPLC/UHPLC system with autosampler
- Target analytical column and a new guard column (if available)
- System suitability standard mixture
- Freshly prepared mobile phases with consistent additives
4.2.2 Procedure
- System Suitability Check: Inject the system suitability standard at the beginning of the sequence. Note the retention times of key peaks.
- Pressure and Leak Check:
- Monitor the system pressure for stability. A lower-than-normal but stable pressure can indicate a leak [113].
- Perform the system's built-in dynamic leak tests, focusing on the autosampler's injector module, as a leaking needle seal is a common culprit for RT shifts without obvious pressure drops [113].
- Mobile Phase and Additive Consistency:
- Ensure that the mobile phase reservoirs contain fresh, properly prepared solvents.
- For methods where one mobile phase contains an additive (e.g., formic acid) and another does not, residual additive can cause pH instability. Prime and flush all lines thoroughly with the final mobile phase composition, or modify the method to include a low concentration of a compatible additive in all mobile phases to stabilize pH [114].
- Column Integrity Test:
- If the above steps fail, the column may be degraded. Replace the column with a new one from the same batch or a new guard column.
- Re-inject the system suitability standard. A return to stable, expected retention times confirms column degradation as the cause [115].
- Temperature Stabilization: Ensure the column compartment temperature is set and stable. For methods at "ambient" temperature, consider controlling the temperature to eliminate fluctuations.
Workflow Visualization
The following diagram illustrates a systematic decision-making process for troubleshooting these chromatographic issues.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and materials critical for maintaining a robust UHPLC system and preventing the issues discussed.
Table 3: Essential Research Reagent Solutions for UHPLC Method Robustness
| Item | Function/Application | Key Considerations |
|---|---|---|
| High-Purity Solvents (HPLC Grade) | Mobile phase components. | Low UV absorbance, especially at shorter wavelengths (< 220 nm). Acetonitrile is often preferred over methanol for low-wavelength work [108]. |
| Volatile Mobile Phase Additives | Modifying pH and improving peak shape for ionizable analytes. | Formic acid and acetic acid are common for MS compatibility. Use consistent, low concentrations (e.g., 0.1%) across all mobile phases to prevent pH instability [114]. |
| In-line Degasser | Removing dissolved gases from the mobile phase. | Prevents bubble formation in the detector flow cell, which is a major cause of baseline noise and spikes [111] [108]. |
| Static Mixer | Ensuring homogeneous mixing of mobile phases before the column. | Critical for minimizing baseline disturbances in gradient methods, especially with additives like TFA [111] [108]. |
| Seal and Valve Maintenance Kit | Routine replacement of wearing components. | Includes needle seals, pump seals, and rotor seals. A leaking needle seal is a documented cause of retention time shifts [113]. |
| System Suitability Standard Mix | Verifying system performance before analytical runs. | A mixture of analytes that tests efficiency, retention, and S/N. Failure to meet criteria indicates a need for troubleshooting [115]. |
Validation and Selection: Ensuring Data Integrity and Choosing the Right Tool
Key Validation Parameters for UFLC and Spectrophotometric Methods
Within pharmaceutical analysis, the choice of analytical technique is pivotal, balancing factors such as sensitivity, selectivity, speed, and cost. Ultra-fast liquid chromatography (UFLC), particularly when coupled with tandem mass spectrometry (MS/MS), represents a high-performance platform for complex analyses. In contrast, spectrophotometry offers a robust, cost-effective alternative for simpler assays. This application note, framed within broader research comparing these techniques, delineates the key validation parameters and provides standardized protocols for each method, supporting researchers and drug development professionals in making informed analytical decisions.
The validation of analytical procedures ensures that their results are reliable, reproducible, and suitable for their intended purpose. The parameters and typical acceptance criteria for UFLC and spectrophotometric methods are summarized in the table below.
Table 1: Key Validation Parameters for UFLC and Spectrophotometric Methods
| Validation Parameter | UFLC-MS/MS Method (as applied to antibiotic analysis) | UV-Vis Spectrophotometric Method (as applied to drug quantification) |
|---|---|---|
| Linearity & Range | 2.0–1000.0 ng/mL [116] | 0.50–3.00 µg/mL for ClAlPc; RIF in various matrices [117] [118] |
| Precision | RSD: 0.56% to 3.5% [116] | RSD: 0.58% to 4.80% for ClAlPc; %RSD 2.06% to 13.29% for RIF [117] [118] |
| Accuracy | Recovery: 57% to 85% [116] | % Recovery: 98.9% to 102.7% for ClAlPc; %RE -11.62% to 14.88% for RIF [117] [118] |
| Limit of Detection (LOD) | Not explicitly stated, but method is highly sensitive [116] | 0.09 µg/mL for ClAlPc; ~0.25-0.49 µg/mL for RIF [117] [118] |
| Limit of Quantification (LOQ) | The lower limit of the linear range (2.0 ng/mL) serves as the LOQ [116] | 0.27 µg/mL for ClAlPc [117] |
| Specificity | High specificity achieved via MRM transitions on MS/MS [116] | Demonstrated selectivity in the presence of common excipients [117] |
| Analysis Speed | Total run time: 2.5 minutes [116] | Generally very fast, with minimal sample preparation [117] [118] |
Detailed Experimental Protocols
Protocol for UFLC-MS/MS Analysis of Antibiotics in Water Samples
This protocol is adapted from a study analyzing 11 antibiotics in pharmaceutical wastewater [116].
Research Reagent Solutions
Table 2: Essential Reagents and Materials for UFLC-MS/MS Antibiotic Analysis
| Item | Function / Specification |
|---|---|
| Analytical Standards | High-purity (>99%) antibiotics (e.g., ceftazidime, ciprofloxacin) for calibration [116] |
| Strata X Cartridge | Mixed-mode reversed-phase/cation-exchange solid-phase extraction (SPE) cartridge (33 µm, 30 mg/1cc) for sample clean-up and analyte concentration [116] |
| UFLC-MS/MS System | Instrumentation consisting of an ultra-fast liquid chromatography system coupled to a triple quadrupole mass spectrometer (e.g., Shimadzu UFLC with Sciex API 4000) [116] |
| Analytical Column | Inertsil ODS C18 column (50 mm × 4.6 mm, 5 µm particle size) for chromatographic separation [116] |
| Mobile Phase A | 10 mM Ammonium formate buffer [116] |
| Mobile Phase B | Methanol or Acetonitrile (HPLC-grade) [116] |
Procedure
- Sample Preparation: Collect water samples (influent, effluent, surface, or groundwater) in sterile polypropylene bottles. Transport and store in a cold chain. Centrifuge if necessary to remove particulates [116].
- Solid-Phase Extraction (SPE):
- Condition the Strata X cartridge with methanol followed by ultrapure water.
- Load a known volume of the water sample onto the cartridge.
- Wash with a suitable solvent to remove impurities.
- Elute the target antibiotics with a solvent like methanol or a methanol/acetonitrile mixture [116].
- Chromatographic Separation:
- Column Oven Temperature: 40 °C
- Auto-sampler Cooler Temperature: 15 °C
- Injection Volume: 10 µL
- Mobile Phase: Utilize a binary gradient with Mobile Phase A (10 mM ammonium formate buffer) and Mobile Phase B (methanol or acetonitrile).
- Flow Rate: 0.5 mL/min
- Total Run Time: 2.5 minutes [116].
- Mass Spectrometric Detection:
- Ion Source: Electrospray Ionization (ESI)
- Detection Mode: Multiple Reaction Monitoring (MRM)
- Ion Transitions: Monitor specific precursor ion > product ion transitions for each antibiotic (e.g., 235.1/105.9 to 711.5/467.9 m/z) [116].
- Data Analysis: Quantify antibiotics using external standard calibration based on peak areas, with a linear range of 2.0–1000.0 ng/mL [116].
Protocol for UV-Vis Spectrophotometric Analysis of Active Compounds
This protocol synthesizes methodologies for quantifying compounds like chloroaluminum phthalocyanine (ClAlPc) and Rifampicin (RIF) [117] [118].
Research Reagent Solutions
Table 3: Essential Reagents and Materials for UV-Vis Spectrophotometric Analysis
| Item | Function / Specification |
|---|---|
| Reference Standard | High-purity compound of interest (e.g., ClAlPc, Rifampicin) for calibration curve [117] [118] |
| Solvent / Medium | Appropriate solvent (e.g., PBS at pH 7.4 or 5.0) or biological matrix (e.g., plasma, tissue homogenate) for sample dissolution/dilution [118] |
| UV-Vis Spectrophotometer | Instrument capable of measuring absorbance at specified wavelengths [117] [118] |
| Cuvettes / Microplates | Disposable or quartz cuvettes for sample holding during measurement |
Procedure
- Standard Solution Preparation: Precisely weigh and dissolve the reference standard in the appropriate solvent (e.g., PBS) to prepare a stock solution. Subsequently, perform serial dilutions to create standard solutions covering the desired concentration range (e.g., 0.50–3.00 µg/mL for ClAlPc) [117] [118].
- Sample Preparation: For pharmaceutical formulations (nanocarriers) or biological matrices (plasma, brain tissue), appropriate pre-treatment such as dilution, filtration, or extraction may be required to ensure the analyte is in solution and within the linear range of the method [117] [118].
- Absorbance Measurement:
- Calibration and Calculation:
- Construct a calibration curve by plotting the absorbance versus the concentration of the standard solutions.
- Determine the regression equation (e.g., Absorbance = Slope × Concentration + Intercept) and the correlation coefficient (r). A value of >0.999 indicates excellent linearity [117] [118].
- Calculate the concentration of the analyte in unknown samples using the regression equation.
The selection between UFLC and spectrophotometric methods is dictated by the analytical problem's specific requirements. UFLC-MS/MS is unequivocally superior for multicomponent analysis at trace levels in complex matrices, such as the simultaneous quantification of multiple antibiotic residues, offering unparalleled specificity, sensitivity, and speed [116]. For single-component analysis where the analyte is present in higher concentrations and the matrix is less interfering, UV-Vis spectrophotometry remains a highly viable, cost-effective, and simple option, capable of delivering fully validated results compliant with ICH guidelines [117] [118]. Understanding these key validation parameters and their associated protocols enables scientists to optimally leverage the strengths of each technique in pharmaceutical research and development.
In the context of discriminating between ultra-fast liquid chromatography and spectrophotometric methods, assessing the selectivity and specificity of an analytical procedure is paramount. A method's selectivity describes its ability to measure the analyte accurately and specifically in the presence of potential interferents, such as impurities, degradation products, or matrix components. The central challenge to achieving this is the phenomenon of co-elution, where two or more compounds do not separate chromatographically, and its consequential spectral overlap, where the signals of these compounds become intertwined [119]. In complex biological or pharmaceutical mixtures, this overlap can severely compromise data accuracy, leading to incorrect quantification and misidentification. This article details application notes and protocols for identifying, addressing, and validating methods against these critical challenges, with a specific focus on applications in drug development.
Computational Strategies for Peak Separation
When chemical or technical solutions for complete chromatographic separation are impractical, computational peak deconvolution becomes an effective strategy. These methods are particularly vital for large-scale experiments with numerous samples [119].
Method 1: Clustering-Based Peak Separation
This method separates convolved fragments of chromatograms into groups of peaks with similar shapes.
Protocol: Clustering for Peak Deconvolution
- Data Pre-processing: Begin by normalizing the raw chromatographic data by the mass of the sample. Follow this by baseline removal and retention time alignment to correct for shifts between runs [119].
- Peak Detection: Apply a peak detection algorithm to identify regions of interest within each chromatogram. For data with low noise, this can be performed without smoothing the second derivative [119].
- Hierarchical Clustering: Within the detected peak regions, use hierarchical clustering with a large number of bootstrap samples (e.g., 1000) to group chromatograms based on the shape of their peaks. The algorithm may initially group peaks into many clusters (e.g., 1 to 18) [119].
- Cluster Joining Algorithm: Apply a specific algorithm to logically join peaks from different clusters. This final step allows for the definitive separation of a single peak or the resolution of a double peak into its two constituent compounds [119].
Method 2: Functional Principal Component Analysis (FPCA)
FPCA does not explicitly separate peaks but provides an optimal, multidimensional representation by detecting sub-peaks with the greatest variability.
Protocol: FPCA for Peak Representation
- Pre-processing: Perform the same pre-processing steps as in Method 1: normalization, baseline removal, and retention time alignment [119].
- Define Basis Functions: Assume a standard peak consists of a fixed number of retention time points (e.g., ~60 points). Use a set of basis functions, such as 6 B-spline functions of order 3, to represent the chromatographic data [119].
- Apply FPCA: Functional Principal Component Analysis is applied to the data. This technique identifies the components within the convolved peaks that account for the most significant variability across different chromatograms [119].
- Interpret Results: A key advantage of FPCA is that it highlights peaks with different areas between experimental variants (e.g., control vs. treatment), which is crucial for comparative untargeted metabolomics. This helps preserve biologically relevant differences that might be lost with other methods [119].
The following workflow illustrates the application of these two computational methods for separating co-eluted peaks:
Experimental Protocol: Validating a UPLC Method for Topical Creams
The following detailed protocol validates the selectivity and specificity of an Ultra-Performance Liquid Chromatography (UPLC) method for quantifying active ingredients in a complex matrix, using Nystatin (Nys) and Triamcinolone Acetonide (TA) in topical cream as a model [89].
Materials and Instrumentation
- Analytical Standards: Nys and TA procured from a certified supplier (e.g., E-Merck) [89].
- Mobile Phase: UPLC-grade solvents, including Tetrahydrofuran, Acetonitrile, and Methanol. Water should be obtained from a Milli-Q system [89].
- Instrumentation: Ultra-Performance Liquid Chromatography (UPLC) system. For in-vitro release testing, Franz diffusion cell instrumentation is required [89].
- Membrane: A 25 mm, 0.45 µm Nylon membrane for the Franz diffusion cell [89].
- Receptor Medium: A 50:50 (v/v) mixture of water and tetrahydrofuran [89].
In-Vitro Release Testing (IVRT) and Sample Preparation
- Membrane Preparation: Soak the nylon membrane in the receptor medium and carefully place it over the bottom cavity of the Franz diffusion cell's sample chamber [89].
- Sample Application: Apply approximately 300 mg of the topical cream sample onto the membrane. Spread it evenly with a spatula to fill the entire cavity of the sample chamber [89].
- Diffusion Parameters: Maintain the receptor medium temperature at 32.0° ± 1.0 °C and set the stirring speed to 500 rpm [89].
- Sample Collection: Withdraw a specified volume of the receptor medium at predetermined time intervals (e.g., 1, 2, 3, 4, 5, and 6 hours). Replace the withdrawn volume with fresh, preheated media at each interval [89].
UPLC Analysis Conditions
- Detection Wavelength: 304 nm for Nys and 254 nm for TA [89].
- Mobile Phase: Utilize an aqueous phase (e.g., 0.1% orthophosphoric acid) and an organic phase (e.g., acetonitrile) in a gradient elution program optimized for separating Nys and TA [89].
- Column: A suitable UPLC column, such as a reverse-phase C18 column with sub-2µm particles [89] [120].
Validation Parameters and Acceptance Criteria
The method should be validated according to International Council for Harmonisation (ICH) guidelines Q2(R2). The table below summarizes key validation parameters and typical acceptance criteria for a method like this [89]:
Table 1: UPLC Method Validation Parameters and Criteria
| Validation Parameter | Experimental Design & Acceptance Criteria |
|---|---|
| Specificity/Selectivity | No interference from blank receptor medium or cream excipients at the retention times of Nys and TA. |
| Linearity & Range | Linearity for TA: 0.65–31.93 µg/mL; Nys: 17.67-863.27 IU/mL. Coefficient of determination (R²) ≥ 0.999 [89]. |
| Precision | Relative Standard Deviation (RSD) for repeatability and intermediate precision < 5.0% [89]. |
| Accuracy | Recovery rates within acceptable ranges (e.g., 77-160% as demonstrated) [89]. |
| Robustness | Method resistant to small, deliberate variations in dose amount, receptor media composition, stirring speed, and temperature [89]. |
This workflow outlines the key stages of method development and validation, from sample preparation through to the final analytical report:
Advanced Techniques and Visualization Tools
Two-Dimensional Liquid Chromatography (2D-LC)
For samples where single-dimension chromatography is insufficient, 2D-LC provides a powerful solution. Multiple Heart-Cutting (mLC-LC) is ideal for analyzing a limited number of known, hard-to-separate impurities in a sample, transferring specific regions from the first to the second dimension [120]. Selective Comprehensive (sLCxLC) mode collects multiple fractions across a region of the 1D separation, preserving relative retention information and improving resolution of partially overlapped peaks, which is highly effective for peak purity analysis [120].
Data Visualization for Quality Assessment
Advanced visualization is critical for assessing data quality and detecting subtle spectral features. Pseudocolor plots can visualize liquid chromatography-mass spectrometry (LC/MS) data by mapping retention time (x-axis), m/z (y-axis), and a third parameter like intensity or variance (z-axis as color) [121]. These plots help identify isotope clusters and in-source fragmentation. To evaluate replicate quality, plot the Relative Maximum Difference from the Mean (RMDM), a variance metric more sensitive to outliers than the coefficient of variation (CV), revealing biases in intensity values [121]. Open-source toolkits built with Python can also parse and visualize MS data via interactive dashboards, aiding in quality control monitoring [122].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Chromatographic Method Development and Validation
| Item | Function & Application |
|---|---|
| UPLC System | Provides high-pressure separations for improved resolution and speed compared to HPLC, essential for ultra-fast methods [89] [42]. |
| Franz Diffusion Cell | Standard apparatus for conducting In-Vitro Release Testing (IVRT) of topical formulations, simulating release through a membrane [89]. |
| Sub-2µm Particle Columns | UPLC columns packed with very fine particles are the cornerstone of modern ultra-fast, high-resolution chromatography [89] [120]. |
| Orthogonal 2D-LC Columns | Using two columns with different separation mechanisms (e.g., reversed-phase and chiral) is key to resolving co-eluting peaks with high selectivity [120]. |
| Solid Phase Extraction (SPE) Cartridges | For sample clean-up and pre-concentration of analytes from complex matrices, improving method sensitivity and reliability [42]. |
| Pseudocolor Plot Software | Customizable software (e.g., using R and Python) for visualizing complex LC/MS datasets to identify patterns and assess data quality [121]. |
Successfully navigating the challenges of co-elution and spectral overlap requires a multi-faceted approach. This article has outlined practical computational and experimental protocols, emphasizing rigorous validation per ICH guidelines. The choice between advanced chromatographic techniques like UPLC or 2D-LC and simpler spectrophotometric methods hinges on the required level of selectivity and specificity. For drug development professionals, employing these detailed strategies and tools is critical for generating reliable, high-quality analytical data that ensures product efficacy, safety, and quality.
Determining Limit of Detection (LOD) and Limit of Quantification (LOQ)
In the field of analytical chemistry, particularly within pharmaceutical development and validation, the determination of a method's sensitivity is paramount. The Limit of Detection (LOD) and Limit of Quantification (LOQ) are two fundamental performance characteristics that define the lowest concentrations of an analyte that can be reliably detected and quantified, respectively [123]. Within the context of ultra-fast liquid chromatography (UFLC) versus spectrophotometric method discrimination research, understanding and accurately determining these parameters allows for a meaningful comparison of the fundamental capabilities of each technique. This document provides detailed application notes and protocols for determining LOD and LOQ, framing them within the practical needs of researchers and drug development professionals.
Theoretical Foundations of LOD and LOQ
Definitions and Relationship to Analytical Techniques
The Limit of Blank (LoB) is defined as the highest apparent analyte concentration expected to be found when replicates of a blank sample containing no analyte are tested. It essentially describes the "noise" of the method [123]. The Limit of Detection (LOD), sometimes called the detection limit (DL), is the lowest analyte concentration that can be reliably distinguished from the LoB, though not necessarily quantified with precise accuracy [123] [124]. In practical terms, it is the level at which an analyst can be confident that a peak is present. The Limit of Quantification (LOQ), or quantitation limit (QL), is the lowest concentration at which the analyte can not only be reliably detected but also quantified with acceptable accuracy and precision, meeting predefined goals for bias and imprecision [123].
The relative positions of these limits differ between techniques. In UFLC, with its sharper peaks and improved signal-to-noise ratios, these limits are often found at lower concentrations compared to conventional HPLC or spectrophotometric methods [125] [8]. For UFLC, the LOQ may be much closer to the LOD due to superior precision at low concentrations, whereas in spectrophotometry, the gap is typically wider.
Regulatory Context and Method Selection
The International Council for Harmonisation (ICH) guideline Q2(R1) provides a standardized framework for the validation of analytical procedures, including the determination of LOD and LOQ [124]. The selection of an appropriate determination method is crucial, as it must be "fit for purpose," aligning with the requirements of the analytical procedure and its intended use [123]. The table below compares the three primary approaches recognized by ICH.
Table 1: Comparison of ICH Q2(R1) Methods for Determining LOD and LOQ
| Method | Basis of Determination | LOD Calculation | LOQ Calculation | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Visual Evaluation | Visual assessment of chromatogram or spectrum | Lowest concentration producing a detectable signal | Lowest concentration producing a quantifiable signal | Simple, quick | Highly subjective, operator-dependent |
| Signal-to-Noise (S/N) | Instrumental measurement of analyte signal vs. baseline noise | Concentration yielding S/N ≈ 2:1 or 3:1 | Concentration yielding S/N ≈ 10:1 | Instrument-derived, less subjective | Sensitive to baseline stability, arbitrary S/N criteria |
| Standard Deviation of Response & Slope | Statistical analysis of calibration curve | ( LOD = \frac{3.3 \sigma}{S} ) | ( LOQ = \frac{10 \sigma}{S} ) | Robust statistical basis, objective | Relies on a well-defined, linear calibration curve at low levels |
Protocols for Determining LOD and LOQ
The following protocols are designed for high-performance liquid chromatography (HPLC) and UFLC systems but can be adapted for spectrophotometric methods.
Protocol A: Determination Based on Standard Deviation and Calibration Curve Slope
This method is considered scientifically robust and is highly recommended for method validation [124].
Experimental Procedure
- Preparation of Standards: Prepare a minimum of five standard solutions at concentrations expected to be in the low, linear range of the assay, including one near the expected LOQ.
- Instrumental Analysis: Analyze each standard solution in triplicate using the developed chromatographic or spectrophotometric method. For UFLC, ensure the flow path (injector, tubing, detector flow cell) is optimized for the reduced column volumes to avoid peak broadening that can impact sensitivity [126].
- Data Collection: Record the analytical response (e.g., peak area, height, or absorbance).
- Linear Regression Analysis: Perform a linear regression analysis on the mean response versus concentration data. From the regression output, obtain the slope (S) of the calibration curve and the standard error of the regression (σ) or the standard deviation of the y-intercept [124].
- Calculation:
- ( LOD = \frac{3.3 \times \sigma}{S} )
- ( LOQ = \frac{10 \times \sigma}{S} )
- Verification: The ICH mandates that calculated LOD and LOQ values must be experimentally verified. Prepare and analyze a minimum of six replicates at the calculated LOD and LOQ concentrations. For LOD, the peak should be reliably detectable in all replicates. For LOQ, the method should demonstrate a precision (relative standard deviation, RSD) of ≤ 20% and an accuracy (bias) of ±20% [124].
Example Calculation from Empirical Data
The workflow below outlines the steps of this protocol, culminating in a sample calculation based on hypothetical data.
Table 2: Example LOD/LOQ Calculation from Calibration Data (Hypothetical UPLC-MS Data)
| Concentration (ng/mL) | Peak Area 1 | Peak Area 2 | Peak Area 3 | Mean Peak Area |
|---|---|---|---|---|
| 0.5 | 1250 | 1310 | 1190 | 1250 |
| 1.0 | 2450 | 2550 | 2350 | 2450 |
| 2.0 | 5050 | 4950 | 4850 | 4950 |
| 5.0 | 12400 | 12550 | 12300 | 12417 |
| 10.0 | 25100 | 24900 | 24800 | 24933 |
| Regression Parameter | Value | |||
| Slope (S) | 2495.2 | |||
| Standard Error (σ) | 182.4 | |||
| LOD | ( \frac{3.3 \times 182.4}{2495.2} = 0.24 \text{ ng/mL} ) | |||
| LOQ | ( \frac{10 \times 182.4}{2495.2} = 0.73 \text{ ng/mL} ) |
Protocol B: Determination Based on Signal-to-Noise Ratio
This method is commonly used in chromatographic techniques and is practical for direct comparison of instrument performance.
Experimental Procedure
- Preparation of Low-Concentration Standard: Prepare an analyte standard at a concentration that produces a signal 5 to 10 times the baseline noise.
- Chromatographic Analysis: Inject the standard solution and record the chromatogram.
- Measurement:
- Measure the peak height (H) of the analyte from the baseline.
- In a blank region of the chromatogram near the analyte peak, measure the peak-to-peak noise (N) over a representative distance.
- Calculation of S/N: Calculate the signal-to-noise ratio as ( S/N = H / N ).
- Extrapolation: The LOD is the concentration that yields an S/N of 3:1. The LOQ is the concentration that yields an S/N of 10:1 [124]. These values can be extrapolated from the measured data:
- ( LOD = \frac{3}{(Measured\ S/N)} \times (Standard\ Concentration) )
- ( LOQ = \frac{10}{(Measured\ S/N)} \times (Standard\ Concentration) )
- Verification: As with Protocol A, verify the extrapolated LOD and LOQ by analyzing replicate samples at those concentrations.
Application in UFLC vs. HPLC and Spectrophotometry
The advantages of UFLC, which utilizes columns packed with sub-2 μm particles, directly impact LOD and LOQ. The sharper, narrower peaks produced by UFLC result in higher signal intensity (peak height) for a given concentration, thereby improving the signal-to-noise ratio [125] [8]. This often leads to lower LOD and LOQ values compared to conventional HPLC.
Table 3: Comparison of LOD/LOQ for Vitamin C Determination by HPLC and UPLC [125] [8]
| Parameter | HPLC Method | UPLC Method | Improvement with UPLC |
|---|---|---|---|
| LOD | 0.049 μg/mL | 0.024 μg/mL | 2.0-fold (50% lower) |
| LOQ | 0.149 μg/mL | 0.073 μg/mL | 2.0-fold (51% lower) |
| Analysis Time | 15 min | 6 min | 60% reduction |
| Eluent Consumption | Higher | Lower | More environmentally friendly |
In contrast, spectrophotometric methods generally have higher LOD and LOQ values than chromatographic techniques due to less selectivity and greater potential for matrix interference. The fundamental difference lies in the "analyte focusing" capability of chromatography, which separates the analyte from background interferences, thereby enhancing the effective S/N ratio at the detector.
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for LOD/LOQ Determination
| Item | Function/Description | Example in Context |
|---|---|---|
| High-Purity Analytical Standards | Certified reference material of the target analyte with known purity and concentration. Essential for preparing accurate calibration standards. | MC-LR standard for toxin analysis [127]; Ascorbic acid for vitamin C assay [125]. |
| Appropriate Blank Matrix | A sample that is as close as possible to the real sample but devoid of the analyte. Used to determine LoB and assess matrix effects. | Blank urine for diuretic analysis [128]; drug-free hair samples [128]. |
| LC-MS Grade Solvents | High-purity solvents (water, methanol, acetonitrile) with minimal UV absorbance and low residue. Critical for low background noise in sensitive detection. | Used in UPLC-MS/MS for diuretics to prevent ion source contamination and high background [128]. |
| Mobile Phase Additives | Acids (e.g., formic acid) or buffers (e.g., ammonium formate) to optimize ionization in MS detection or control retention in chromatography. | 0.1% formic acid in UPLC-MS/MS to promote protonation [128]; 0.05% formic acid for MC-LR analysis [127]. |
| Sub-2μm UPLC Columns | Short, narrow-bore columns packed with small particles (<2 μm) for high-resolution, high-speed separations with reduced dispersion. | ACQUITY UPLC BEH C18 column (1.7 μm, 50 mm x 2.1 mm) for fast analysis of diuretics [128]. |
| Sample Filtration Membranes | Low-binding membranes to remove particulates from samples without adsorbing the analyte, which could lead to inaccurate low concentration results. | Evaluation of various membranes (NY, MCE, PTFE, PES) for MC-LR analysis to avoid adsorption losses [127]. |
Comparative Case Study: MC-LR Analysis by UPLC-MS vs. HPLC-VWD
A direct comparison of UPLC-MS and HPLC with a variable wavelength detector (VWD) for the analysis of Microcystin-LR (MC-LR) highlights the technique-dependent nature of LOD/LOQ. The UPLC-MS method, leveraging the sensitivity and selectivity of mass spectrometry, demonstrated a LOD of 0.03–0.05 μg L⁻¹ and an LOQ of 0.08 μg L⁻¹. In contrast, the HPLC-VWD method, relying on UV absorption, showed a LOD of 0.6 μg L⁻¹ and an LOQ of 1.0 μg L⁻¹ [127]. This represents a 12-20 fold improvement in sensitivity for the UPLC-MS method.
The workflow below illustrates the decision-making process for selecting the appropriate analytical method based on the expected concentration range, as demonstrated in the MC-LR study.
This case study underscores a critical consideration: while UPLC-MS is superior for trace analysis, HPLC-VWD remains a robust and cost-effective choice for analyzing high-concentration samples, avoiding potential ion source contamination in the mass spectrometer [127].
Accurate determination of LOD and LOQ is a critical component of analytical method validation. The choice of calculation method—whether based on calibration curve statistics, signal-to-noise, or visual evaluation—should be guided by the technique's nature and regulatory requirements. As demonstrated, UFLC methodologies, particularly when coupled with mass spectrometric detection, consistently offer superior sensitivity with lower LOD and LOQ values compared to conventional HPLC and spectrophotometric methods. This enhanced performance enables more precise and accurate quantification of analytes at trace levels, a decisive factor in demanding applications within pharmaceutical research, food safety, and environmental monitoring.
Within pharmaceutical development, the selection of an analytical technique is a critical decision that balances analytical performance with practical and economic constraints. This application note provides a detailed comparative analysis of Ultra-Fast Liquid Chromatography (UFLC) and UV Spectrophotometry across the key parameters of speed, sensitivity, specificity, and cost of ownership. Framed within broader research on method discrimination, this document delivers structured data and detailed experimental protocols to guide researchers and drug development professionals in making informed, application-driven choices.
The following table summarizes a quantitative comparison of the core characteristics of UFLC and UV Spectrophotometry.
Table 1: Comparative Analysis of UFLC and UV Spectrophotometry
| Parameter | Ultra-Fast Liquid Chromatography (UFLC) | UV Spectrophotometry |
|---|---|---|
| Speed | Run times of 1.30 to 2.35 minutes for online process monitoring [129]. ~10x faster than conventional HPLC [130]. | Samples can be processed in ~5-10 minutes with minimal preparation [131]. |
| Sensitivity | Mass spectrometry detection can offer up to 1000x lower detection levels for some analytes compared to spectrophotometry [131]. | Limited sensitivity for trace-level analytes or early disease biomarkers; may not detect low-concentration components [131]. |
| Specificity | High specificity from compound separation and advanced detection (e.g., MS). Can resolve and quantify individual components in complex mixtures like Polysorbate 20 [73]. | Low specificity for mixtures due to spectral overlap; requires mathematical processing (e.g., derivative, ratio spectra) for resolution [132]. |
| Capital Cost | High; systems can cost upwards of $35,000, with columns alone at ~$1,000 [130]. | Low; instrumentation is relatively inexpensive [131]. |
| Operational Cost | High ongoing costs for solvents, consumables (columns, vials, filters), and maintenance [130]. | Low; minimal solvent use and fewer consumables [132]. |
Detailed Experimental Protocols
Protocol 1: Ultra-Fast Liquid Chromatography for Real-Time Process Monitoring
This protocol, adapted from cutting-edge Process Analytical Technology (PAT) applications, is designed for monitoring critical quality attributes during biotherapeutic purification [129].
I. Objectives
- To separate and quantify product variants in complex biopharmaceutical samples.
- To achieve sampling cycle times of under 2.5 minutes for real-time decision-making.
II. Materials and Reagents
- UHPLC System: A suitable ultra-high-performance liquid chromatography system.
- Columns: Columns appropriate for the selected separation mode.
- Mobile Phase: Prepared from HPLC-grade solvents and filtered through a 0.22 µm membrane.
- Samples: In-process samples from downstream bioprocessing.
III. Instrumentation and Conditions
- System: Novel online LC setup configured for high-speed analysis.
- Configuration: Two-pump, two-column system for parallel column conditioning and analysis.
- Flow Rate: Optimized for high flow (e.g., 1.0-2.0 mL/min).
- Gradient: Fast, steep organic gradient.
- Detection: UV detector.
- Temperature: Column oven set to 40-60°C to reduce backpressure and improve kinetics.
IV. Procedure
- System Setup: Install and equilibrate the analytical column in the online LC setup.
- Method Programming: Develop a fast gradient method. The key innovation is programming the system to perform column loading, washing, and equilibration on one column offline, in parallel with the analytical gradient run on the other column.
- Sample Preparation: Filter in-process samples via centrifugation and 0.22 µm filtration.
- Analysis: Inject the prepared sample. The tandem direct injection workflow eliminates traditional method overhead, enabling cycle times between 1.30 and 2.35 minutes.
- Data Analysis: Integrate peaks and quantify analytes of interest using a chromatography data system (CDS).
Protocol 2: Spectrophotometric Analysis of a Drug Mixture using Ratio Difference Method
This protocol details the analysis of a binary drug mixture (Paracetamol and Domperidone) where significant spectral overlap occurs, utilizing a ratio difference method for resolution [132].
I. Objectives
- To simultaneously determine the concentration of two drugs with overlapping UV spectra in a laboratory-made tablet.
- To assess the greenness of the analytical method.
II. Materials and Reagents
- Drug Standards: Paracetamol (PAR) and Domperidone (DOM).
- Solvent: HPLC-grade Methanol.
- Apparatus: Double-beam UV-Vis spectrophotometer with 1-cm quartz cells.
III. Instrumentation and Conditions
- Instrument: Thermo Spectronic Helios Alpha or equivalent.
- Wavelength Range: 200-400 nm.
- Data Processing: Software capable of storing spectra and performing mathematical transformations (e.g., deriving ratio spectra).
IV. Procedure
- Standard Solution Preparation
- Prepare 1000 µg/mL stock solutions of PAR and DOM separately in methanol.
- Dilute aliquots to create working standard solutions in the ranges of 3–70 µg/mL for PAR and 2.5–15 µg/mL for DOM.
Sample Solution Preparation
- Weigh and grind 20 tablets. Weigh a portion of powder equivalent to 10 mg of DOM.
- Extract the powder with 20 mL methanol, sonicate for 30 minutes, and filter into a 100 mL volumetric flask. Dilute to volume with methanol.
- Perform further dilutions with methanol to bring the concentration within the working range.
Analysis by Ratio Difference Method
- For PAR Quantification:
- Scan and store the absorption spectra of all PAR standard and sample solutions.
- Divide (ratio) each stored spectrum by the spectrum of a 50 µg/mL standard DOM solution (the "divisor").
- Measure the amplitudes of the resulting ratio spectra at 256 nm and 288 nm.
- Plot the difference between these amplitudes (A@256nm - A@288nm) against the corresponding PAR concentrations to create a calibration curve.
- For DOM Quantification:
- Scan and store the absorption spectra of all DOM standard and sample solutions.
- Divide each stored spectrum by the spectrum of a 50 µg/mL standard PAR solution (the "divisor").
- Measure the amplitudes of the resulting ratio spectra at 216 nm and 288 nm.
- Plot the difference between these amplitudes (A@216nm - A@288nm) against the corresponding DOM concentrations.
- Use the respective calibration curves to determine the concentration of PAR and DOM in the sample solutions.
- For PAR Quantification:
Workflow and Logical Pathway Visualization
The following diagram illustrates the core decision-making workflow for selecting between UFLC and UV Spectrophotometry based on analytical requirements and constraints.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and reagents required for the experimental protocols featured in this note.
Table 2: Essential Research Reagent Solutions
| Item | Function / Application | Example / Specification |
|---|---|---|
| HPLC/UHPLC System | Core instrumentation for UFLC separations. | Systems capable of high-pressure operation (e.g., up to 1300 bar); e.g., Agilent Infinity III, Shimadzu i-Series, Thermo Vanquish Neo [30]. |
| C18 Analytical Column | Stationary phase for reverse-phase chromatographic separation. | Agilent TC-C18 (250 mm × 4.6 mm, 5 µm) or equivalent [39]. |
| HPLC-Grade Solvents | Mobile phase components; require high purity to minimize background noise. | Methanol, Acetonitrile, Water (filtered through 0.22 µm membrane) [132] [39]. |
| Drug Standards | Primary reference materials for method development, calibration, and validation. | Certified Reference Standards of analytes (e.g., Repaglinide, Paracetamol, Meloxicam) [132] [39]. |
| UV-Vis Spectrophotometer | Core instrumentation for measuring light absorption by analytes. | Double-beam instrument with 1-cm quartz cells; e.g., Shimadzu 1700, Thermo Helios Alpha [132] [39]. |
| Solid-Phase Extraction (SPE) Kits | Automated sample preparation and cleanup for complex matrices. | Pre-packaged kits with stacked cartridges for specific assays (e.g., PFAS, oligonucleotides) [133]. |
Guidelines for Method Validation in Regulatory Environments
In pharmaceutical development and manufacturing, demonstrating that an analytical method is reliable and fit for its intended purpose is a fundamental regulatory requirement. For researchers comparing advanced techniques like ultra-fast liquid chromatography (UFLC) and spectrophotometric methods, adherence to these validated guidelines is not optional but mandatory for regulatory acceptance. The International Council for Harmonisation (ICH), the U.S. Food and Drug Administration (FDA), and other regulatory bodies provide the foundational frameworks that define the validation parameters and acceptance criteria for analytical procedures [134] [59]. These guidelines ensure that analytical data supporting drug release, stability studies, and regulatory submissions possesses the necessary integrity, accuracy, and reliability to guarantee product quality and patient safety.
The simultaneous recent publication of ICH Q2(R2) and ICH Q14 signifies a significant modernization in the regulatory landscape, moving from a prescriptive, "check-the-box" approach to a more scientific, risk-based lifecycle model [59]. For scientists engaged in method discrimination research, this shift emphasizes that validation is not a one-time event but a continuous process that begins with method development and continues throughout the method's operational life. This article details the core validation parameters, experimental protocols, and practical workflows that researchers must implement to comply with global regulatory standards, with a specific focus on applications within chromatographic and spectroscopic analysis.
Core Regulatory Guidelines and Validation Parameters
Key Global Regulatory Guidelines
Adherence to globally harmonized guidelines streamlines the path from method development to regulatory approval. The following are the three most critical documents for analytical method validation in the pharmaceutical sector.
- ICH Q2(R2): Validation of Analytical Procedures: This is the primary global reference, defining what constitutes a valid analytical procedure [134] [59]. The 2024 revision (Q2(R2)) expands its scope to include modern technologies like UFLC and mass spectrometry and reinforces a science- and risk-based approach to validation. It applies to procedures used for the release and stability testing of commercial drug substances and products [134].
- FDA Analytical Procedures and Methods Validation Guidance: The FDA, as a key ICH member, adopts and implements the harmonized principles [59]. Its guidance expands on the ICH framework, providing detailed recommendations for the U.S. regulatory landscape, with a particular emphasis on method robustness and comprehensive documentation of analytical accuracy [135].
- USP <1225> Validation of Compendial Procedures: This chapter establishes the validation requirements for methods published in the United States Pharmacopeia (USP) [135]. It categorizes analytical procedures into distinct types (e.g., identification tests, assays, impurity tests) and specifies which validation parameters are required for each category.
Essential Validation Parameters
Regulatory guidelines stipulate a set of fundamental performance characteristics that must be evaluated to demonstrate a method is fit-for-purpose. Table 1 summarizes these core parameters, their definitions, and typical acceptance criteria for quantitative assays.
Table 1: Core Validation Parameters for Quantitative Analytical Procedures
| Parameter | Regulatory Definition | Typical Acceptance Criteria & Experimental Approach |
|---|---|---|
| Accuracy | The closeness of agreement between the test result and the true value [59]. | Recovery of 98–102% for drug substance; 98–102% for drug product (from specific matrix). Assessed by spiking a placebo with a known amount of analyte or comparing to a reference standard [59]. |
| Precision | The degree of agreement among individual test results when the procedure is applied repeatedly to multiple samplings of a homogeneous sample [59]. | Repeatability (Intra-assay): RSD ≤ 1.0% for drug substance, ≤ 2.0% for drug product.Intermediate Precision: RSD ≤ 2.0% (incorporates inter-day, inter-analyst, inter-instrument variability). |
| Specificity | The ability to assess the analyte unequivocally in the presence of components that may be expected to be present [59]. | Chromatographic: Baseline resolution of analyte from closest eluting potential interferent (Resolution > 1.5). Spectroscopic: No significant interference at the analyte's detection wavelength. |
| Linearity | The ability of the method to elicit test results that are directly proportional to analyte concentration [59]. | Correlation coefficient (r) > 0.998 [59]. Demonstrated across the specified range using a minimum of 5 concentration levels. |
| Range | The interval between the upper and lower concentrations of analyte for which suitable levels of linearity, accuracy, and precision have been demonstrated [134]. | Defined by the linearity and precision studies, typically from 80% to 120% of the test concentration for assay methods. |
| Limit of Detection (LOD) | The lowest amount of analyte in a sample that can be detected, but not necessarily quantitated [59]. | Signal-to-noise ratio (S/N) ≥ 2 or 3. Evaluated by visual inspection or based on the standard deviation of the response and the slope of the calibration curve. |
| Limit of Quantitation (LOQ) | The lowest amount of analyte in a sample that can be quantitatively determined with suitable precision and accuracy [59]. | S/N ≥ 10. Accuracy and precision at LOQ should be within ±20% and RSD ≤ 20%, respectively. |
| Robustness | A measure of the method's capacity to remain unaffected by small, deliberate variations in method parameters [59]. | System suitability criteria are met when parameters (e.g., mobile phase pH (±0.2), flow rate (±5%), column temperature (±2°C)) are intentionally varied. |
Application in Method Discrimination: UFLC vs. Spectrophotometry
Validation Considerations for Technique Selection
The choice between UFLC and spectrophotometry for a specific analytical task is heavily influenced by the aforementioned validation parameters. A method's intended purpose, defined early in development through an Analytical Target Profile (ATP), dictates which technique is more appropriate [59]. The ATP prospectively summarizes the method's required performance characteristics, such as the required sensitivity, specificity, and throughput.
Specificity is often the most significant differentiator. UFLC, especially when coupled with mass spectrometry (MS) or tandem mass spectrometry (MS/MS), provides superior separation and identification of analytes in complex mixtures [136] [19]. For example, in the analysis of citrus metabolites, LC-MS/MS was able to distinguish 342 primary metabolites and 77 phenolic acids across five citrus varieties, a task far beyond the capability of conventional spectrophotometry [136]. In contrast, a simple UV-spectrophotometric method may lack the specificity to resolve the analyte from excipients, degradation products, or matrix components, leading to inaccurate results.
Sensitivity requirements, defined by the LOD and LOQ, also guide the selection. UFLC-MS/MS methods consistently achieve sub-ng/mL sensitivity, as demonstrated in a bioequivalence study for donepezil where the LOQ was 0.2 ng/mL [19]. Spectrophotometry generally has higher detection limits, making it unsuitable for trace-level impurity profiling or low-dose pharmacokinetic studies.
Case Study: Validation of an UFLC-MS/MS Method
The following protocol outlines the validation of a fast UFLC-MS/MS method, showcasing the application of regulatory guidelines in a practical research setting.
- Objective: To develop and validate a rapid, specific, and sensitive UFLC-MS/MS method for the quantification of an active pharmaceutical ingredient (API) in human plasma to support a bioequivalence study.
- Instrumentation:
- LC System: Ultra-fast liquid chromatography system (e.g., ExionLC AC, Shimadzu i-Series, or Agilent Infinity III) [30] [19].
- MS Detector: Triple quadrupole mass spectrometer (e.g., SCIEX Triple Quad 6500+ or equivalent) with electrospray ionization (ESI) [19].
- Column: High-resolution monolithic C18 column (50 x 4.6 mm) for fast separations [19].
- Experimental Workflow:
Figure 1: Experimental workflow for the validation of a bioanalytical method, outlining the key stages from planning to reporting.
- Detailed Protocol:
- Sample Preparation: Use a simple protein precipitation technique. Transfer 200 µL of plasma into a tube, add 50 µL of internal standard (IS) working solution, and vortex. Precipitate proteins with 500 µL of methanol, vortex mix vigorously, and centrifuge at 3,500 × g for 5 minutes. Transfer the supernatant, dilute with water, and inject into the LC-MS/MS system [19].
- Chromatographic Conditions:
- Mobile Phase: A: 0.1% Formic acid in water; B: 0.1% Formic acid in acetonitrile [19].
- Gradient: 25% B to 80% B over 0.6 minutes, then to 80% B at 1.1 minutes.
- Flow Rate: Multi-stage flow (initially 3 mL/min for 0.1 min, reduced to 1.2 mL/min for separation, then back to 3 mL/min for re-equilibration) [19].
- Column Temperature: Room temperature.
- Injection Volume: 5 µL.
- Run Time: 1.5 minutes [19].
- Mass Spectrometric Detection:
- Ionization Mode: Positive electrospray ionization (ESI+).
- Detection Mode: Multiple Reaction Monitoring (MRM).
- Ion Transitions: Monitor the specific precursor-to-product ion transitions for both the API and the IS [19].
- Source Parameters: Optimize parameters like ion spray voltage, source temperature, and gas settings according to the instrument manufacturer's guidelines.
- Validation Experiments & Data Reporting:
- Specificity: Analyze blank plasma from at least six different sources to demonstrate no interference at the retention times of the API and IS [19].
- Linearity and Range: Prepare a calibration curve with a minimum of six non-zero concentrations. Process and analyze in duplicate. The correlation coefficient (r) should be >0.995 [19]. Table 2 shows an example data structure.
- Accuracy and Precision: Assay QC samples at a minimum of four concentration levels (LLOQ, Low, Medium, High) with at least six replicates per level in a single run (repeatability) and over three different days (intermediate precision). Accuracy should be within 85–115% (100% at LLOQ) and precision (RSD) ≤15% (≤20% at LLOQ) [19].
Table 2: Example Data Structure for Linearity and Precision/Accuracy in an UFLC-MS/MS Method
| Validation Parameter | Concentration Level (ng/mL) | Mean Found (ng/mL) | Accuracy (%) | Precision (RSD, %) |
|---|---|---|---|---|
| Linearity | 0.2 (LLOQ) | 0.21 | 105.0 | - |
| 0.6 | 0.59 | 98.3 | - | |
| 9.0 | 8.91 | 99.0 | - | |
| 20.0 | 19.8 | 99.0 | - | |
| 35.0 | 34.7 | 99.1 | - | |
| 50.0 (ULOQ) | 49.5 | 99.0 | - | |
| Precision & Accuracy (Intra-day) | 0.6 (Low QC) | 0.58 | 96.7 | 4.5 |
| 9.0 (Mid QC) | 8.95 | 99.4 | 2.1 | |
| 35.0 (High QC) | 34.2 | 97.7 | 1.8 |
The Scientist's Toolkit: Essential Reagents and Materials
Successful method validation requires not only a robust protocol but also high-quality, well-characterized materials. The following table lists key reagents and their critical functions.
Table 3: Essential Research Reagent Solutions for Method Validation
| Item | Function & Importance in Validation |
|---|---|
| Analytical Reference Standard | Certified material of known purity and identity. Serves as the benchmark for all quantitative measurements; its quality is foundational to accuracy [19]. |
| Stable Isotope-Labeled Internal Standard (IS) | A deuterated (e.g., Donepezil-d5) or other isotopically labeled version of the analyte. Corrects for variability in sample preparation and ionization efficiency in LC-MS/MS, improving precision and accuracy [19]. |
| HPLC-Grade Solvents & Reagents | High-purity mobile phase components (water, acetonitrile, methanol) and additives (formic acid, ammonium formate). Minimize background noise, prevent system contamination, and ensure reproducible chromatographic performance [19]. |
| Blank Control Matrix | The biological fluid or sample matrix without the analyte (e.g., K2EDTA human plasma). Essential for assessing specificity, matrix effects, and for preparing calibration standards and quality control samples [19]. |
| Quality Control (QC) Samples | Samples with known concentrations of analyte prepared in the control matrix. Used to monitor the performance of the analytical method during validation and in every subsequent run to ensure ongoing reliability [19]. |
Navigating the regulatory guidelines for method validation is a critical competency for scientists developing and implementing analytical methods for drug development. The frameworks provided by ICH, FDA, and USP create a clear, albeit rigorous, pathway to demonstrating that a method—whether UFLC-based or spectrophotometric—is fit for its intended purpose. The paradigm shift towards a science- and risk-based lifecycle approach, as championed by ICH Q2(R2) and Q14, empowers researchers to build quality into their methods from the outset. For method discrimination studies, the validation parameters of specificity, sensitivity, and robustness provide objective criteria for selecting the most appropriate analytical technique. By adhering to the detailed experimental protocols and utilizing a well-characterized toolkit of reagents, researchers can ensure their analytical methods not only meet stringent global regulatory standards but also generate the high-quality data essential for making informed decisions in pharmaceutical development.
The analysis of complex mixtures, such as biological fluids, natural products, and pharmaceutical formulations, presents a significant challenge for researchers and drug development professionals. In these samples, trace-level constituents often coexist with highly abundant compounds, and some of these trace components can possess high activity or toxicity, making their accurate detection and quantification critically important [137]. The primary analytical hurdle in such matrices is the matrix effect, where co-eluting substances can cause severe ion suppression, dramatically affecting method performance in terms of detection capability, selectivity, repeatability, accuracy, and linearity [137]. The presence of highly abundant compositions can mask the detection of lower abundance constituents, which is particularly problematic for samples containing diverse molecules across large orders of magnitude of concentration [137].
While spectrophotometric methods remain popular due to their procedural simplicity, instrument availability, precision, speed, and accuracy [22], they face substantial limitations when dealing with complex mixtures. These difficulties include required larger sample amounts and limitations in recording higher sample concentrations, alongside serious challenges in resolving overlapping bands of analytes and interferences, which make quantitative data analysis complex [22]. In contrast, Ultra-Fast Liquid Chromatography (UFLC) offers superior selectivity and sensitivity for analyzing organic compounds and quantifying isolated substances in mixtures, with advantages including shorter analysis time, increased peak capacity, and reduced consumption of samples and solvents [22]. This application note delineates the specific scenarios where UFLC provides decisive advantages over spectrophotometric methods for complex mixture analysis and trace detection, supported by experimental data and detailed protocols.
Comparative Performance: UFLC vs. Spectrophotometry
Direct comparison studies provide compelling evidence for the superior performance of UFLC in analytical applications requiring high specificity and sensitivity. Research quantifying active pharmaceutical ingredients such as metoprolol tartrate (MET) and repaglinide demonstrates consistent advantages of UFLC-based methodologies.
Table 1: Validation Parameters for MET Determination (UFLC-DAD vs. Spectrophotometry)
| Validation Parameter | UFLC-DAD Method | Spectrophotometric Method |
|---|---|---|
| Specificity/Selectivity | High (Baseline separation of analytes) | Limited (Overlapping spectra in mixtures) |
| Linearity (R²) | >0.999 | >0.999 |
| Precision (% R.S.D.) | <1.50% | <1.50% |
| Accuracy (% Recovery) | 99.71-100.25% | 99.63-100.45% |
| Sample Volume | Low | Larger amounts required |
| Higher Concentration Detection | Effective | Limited |
Table 2: Analytical Method Performance for Repaglinide Quantification
| Parameter | RP-HPLC Method | UV Spectrophotometric Method |
|---|---|---|
| Linearity Range | 5-50 μg/ml | 5-30 μg/ml |
| Regression Coefficient (r²) | >0.999 | >0.999 |
| Precision (% R.S.D.) | <2.0% | <2.0% |
| Mean Recovery | 99.71-100.25% | 99.63-100.45% |
| Limit of Detection (LOD) | Lower | Higher |
| Limit of Quantification (LOQ) | Lower | Higher |
The data reveals that while both methods can be validated to meet regulatory standards, UFLC extends the linear dynamic range and offers lower detection limits, which is crucial for trace analysis [22] [39]. Furthermore, UFLC's superior specificity allows for reliable quantification in the presence of structurally similar compounds and formulation excipients, a challenge that often compromises spectrophotometric analysis [22] [39].
Advanced UFLC Applications in Complex Matrices
Trace Analysis in Natural Products and Herbal Medicines
The analysis of traditional herbal medicines like Dachengqi Decoction (DCQD) exemplifies the power of UFLC when integrated with mass spectrometry. These formulations present formidable challenges due to their complex composition and inconsistencies in traditional preparation processes. Researchers established an integrated approach using UPLC-Q-TOF-MS for qualitative analysis and UFLC-QQQ-MS for quantitative analysis, successfully detecting 190 components and unambiguously identifying 27 compounds [138]. By optimizing parameters including mobile phase composition, gradient, and velocity, the method achieved superior separation of structurally similar substances like aloe-emodin, emodin, and apigenin [138]. This comprehensive profiling enabled the quantification of 19 key ingredients across 10 different formulations, revealing significant differences in component distribution that explain varied therapeutic effects in treating intestinal obstruction and pancreatitis [138].
Targeted Multi-Dimensional Chromatography
For exceptionally complex matrices, targeted multidimensional liquid chromatography (MDLC) provides enhanced resolving power. While conventional two-dimensional methods have been used extensively since the 1970s, recent advances have explored the benefits of adding a third dimension of separation [139]. Using statistical peak overlap theory and advanced models of reversed-phase selectivity, researchers have constructed three-dimensional HPLC systems built on three very different reversed-phase columns [139]. This approach enables excellent separations of target compounds from challenging sample matrices including urban wastewater treatment effluent, human urine, and river water, achieving method detection limits in the low parts-per-trillion range [139]. The system accomplishes this using similar separation conditions for diverse target compound and sample matrix combinations, significantly reducing the normally tedious method development process [139].
High-Component Filtering Strategies
An emerging application of UFLC in complex mixture analysis involves high-component filtering strategies based on heart-cutting techniques. This approach specifically removes high-abundance compounds to enrich trace components, thereby improving their detection [137]. The technique demonstrates that after filtering of high target-components, the detection capacity and the sample loading amount can be considerably increased, enabling the analysis of trace constituents that would otherwise be masked by more abundant compounds [137]. Compared to conventional extraction approaches like solid-phase extraction (SPE), heart-cutting technology offers much higher selectivity by specifically removing interesting ingredients from complex samples [137].
Experimental Protocols
Protocol 1: Quantification of Active Components in Pharmaceutical Tablets
This protocol for metoprolol tartrate (MET) quantification can be adapted for similar pharmaceuticals [22].
- Reagents and Standards: MET reference standard (≥98%), ultrapure water, methanol (HPLC grade).
- Equipment: UFLC system with DAD detector, analytical balance, ultrasonic bath.
- Chromatographic Conditions:
- Column: C18 column (e.g., Agilent ZORBAX SB-C18, 4.6 mm × 250 mm, 5 μm)
- Mobile Phase: Methanol-water or acetonitrile-water in optimized ratios
- Flow Rate: 1.0 mL/min
- Detection: UV at compound-specific λmax (e.g., 223 nm for MET)
- Injection Volume: 10-20 μL
- Sample Preparation:
- Accurately weigh and finely powder 20 tablets.
- Transfer a portion equivalent to 10 mg of active component to a 100 mL volumetric flask.
- Add approximately 30 mL of methanol and sonicate for 15 minutes.
- Dilute to volume with methanol and mix well.
- Filter through a 0.45 μm membrane filter.
- Further dilute with mobile phase to obtain concentrations within the linearity range.
- Method Validation:
- Establish linearity using at least six concentration levels in triplicate.
- Determine precision through repeatability (six injections at 100% test concentration) and intermediate precision (different days, analysts).
- Assess accuracy through standard addition recovery studies at three concentration levels (80%, 100%, 120%).
- Verify specificity by analyzing placebo formulations and forced degradation samples.
Protocol 2: Comprehensive Profiling of Complex Herbal Formulations
This protocol describes the qualitative and quantitative analysis of multi-component herbal preparations like Dachengqi Decoction [138].
- Reagents and Standards: Reference standards for target compounds, methanol (HPLC grade), acetonitrile (HPLC grade), formic acid or acetic acid (high purity), high-purity water.
- Equipment: UFLC system coupled to QQQ-MS, analytical balance, ultrasonic bath, centrifuge.
- Chromatographic Conditions:
- Column: C18 column suitable for UFLC
- Mobile Phase: (A) 0.1% formic acid in water; (B) acetonitrile or methanol
- Gradient Elution: Optimized for target compounds (e.g., 5-60% B over 20-30 minutes)
- Flow Rate: 0.3-0.5 mL/min
- Column Temperature: 35-40°C
- Injection Volume: 1-5 μL
- Mass Spectrometry Conditions:
- Ionization Mode: ESI positive/negative depending on analytes
- Detection Mode: Multiple Reaction Monitoring (MRM) for quantification
- Source Parameters: Optimized for nebulizer gas, drying gas, and source temperature
- Sample Preparation:
- Pulverize herbal material or formulation and accurately weigh a representative sample.
- Extract with appropriate solvent (e.g., methanol-water 7:3) by sonication.
- Adjust to initial weight with extraction solvent.
- Centrifuge and filter supernatant through 0.22 μm membrane filter.
- Dilute with mobile phase if necessary.
- Data Analysis:
- Identify compounds by comparing retention times and MS/MS spectra with reference standards.
- For unknown compounds, use accurate mass measurements and fragmentation patterns.
- Quantify using calibration curves of available reference standards.
Figure 1: UFLC-MS Workflow for Complex Mixtures
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Reagents and Materials for UFLC Analysis of Complex Mixtures
| Item | Function | Application Notes |
|---|---|---|
| HPLC-Grade Methanol & Acetonitrile | Mobile phase components | Low UV cutoff, minimal impurities for sensitive detection |
| High-Purity Water (18 MΩ·cm) | Aqueous mobile phase component | Prepared via Milli-Q or equivalent systems |
| Formic Acid/Acetic Acid (≥96%) | Mobile phase additive | Improves ionization in MS detection and peak shape |
| Ammonium Acetate/Formate | Volatile buffer salts | MS-compatible for pH control |
| Reference Standards | Method development and quantification | High purity (≥98%) for accurate calibration |
| UFLC Columns (C18, phenyl, etc.) | Stationary phases for separation | Different selectivities for various compound classes |
UFLC technology provides an indispensable analytical platform for researchers and drug development professionals working with complex mixtures and trace analysis. The decision to implement UFLC over spectrophotometric methods is justified when analyses require (1) superior specificity in complex matrices, (2) enhanced sensitivity for trace-level components, (3) expanded linear dynamic range, and (4) reliable quantification in the presence of structurally similar compounds. As demonstrated through the presented applications and protocols, UFLC—especially when hyphenated with mass spectrometry—delivers unparalleled capability for characterizing complex samples across pharmaceutical, natural product, and biomedical research domains.
Within pharmaceutical analysis, a central tenet of method selection is aligning the technique's capabilities with the analytical problem. While sophisticated techniques like ultra-fast liquid chromatography (UFLC) and liquid chromatography-mass spectrometry (LC-MS) provide unparalleled separation and specificity for complex mixtures, their deployment for every analysis is neither efficient nor necessary [30] [140]. This application note frames spectrophotometry within this broader methodological context, delineating its definitive advantages in scenarios demanding simplicity, cost-effectiveness, and rapid analysis of high-concentration active pharmaceutical ingredients (APIs). Spectrophotometry, based on the measurement of light absorption by molecules, remains a cornerstone technique in pharmaceutical laboratories for quantitative analysis, dissolution testing, and stability studies [64]. Its principle, governed by the Beer-Lambert Law, offers a direct and robust relationship between analyte concentration and absorbance, making it ideal for targeted, high-throughput assays where its limitations of sensitivity or selectivity are not a hindrance [64] [141].
Spectrophotometry vs. Chromatography: A Strategic Comparison
The choice between spectrophotometry and chromatographic methods is strategic. Advanced chromatographic systems excel at resolving complex mixtures, identifying impurities, and analyzing trace components in challenging matrices like biological fluids [30] [140]. However, these capabilities come with significant operational complexity, higher costs, and longer analysis times. In contrast, modern spectrophotometry, particularly with mathematical manipulation of spectral data, can resolve many overlapping analyses without physical separation, offering a compelling alternative for specific applications [37] [142].
Table 1: Strategic Method Selection: Spectrophotometry vs. Ultra-Fast Liquid Chromatography
| Parameter | Spectrophotometry | Ultra-Fast Liquid Chromatography (UFLC) |
|---|---|---|
| Analytical Principle | Light absorption by molecules [141] | Physicochemical separation followed by detection [30] |
| Key Advantage | Simplicity, speed, and low cost per analysis [64] | High specificity and powerful separation of complex mixtures [140] |
| Typical Analysis Time | Minutes [64] | 2-5 minutes for fast UHPLC methods [140] |
| Instrument Cost | Low to moderate [143] | High |
| Solvent Consumption | Low (often < 10 mL per sample) [37] | Low to moderate (gradient runs) [30] |
| Ideal Use Case | Assay of single APIs or resolved mixtures in formulations; dissolution testing [64] | Multi-component assays; impurity profiling; bioanalysis [64] [140] |
| Throughput for Routine QC | Very High | High |
Key Application Domains for Spectrophotometry
API Assay in Formulations
Spectrophotometry is extensively used for the assay of Active Pharmaceutical Ingredients (APIs) in both bulk and formulated dosage forms like tablets and capsules, providing a rapid and reliable means to ensure correct dosage [64]. For drugs with strong chromophores, UV-Vis spectrophotometry allows for direct quantification with minimal sample preparation.
Dissolution Testing
In dissolution studies, spectrophotometry is the workhorse technique for monitoring the drug release rate from solid dosage forms. Its speed enables high-frequency sampling, which is crucial for generating accurate release kinetics profiles for bioavailability studies [64].
Stability and Impurity Profiling
While less specific than chromatography for low-level impurities, spectrophotometry is highly effective for stability testing. It can track the formation of degradation products under various stress conditions (e.g., heat, light, humidity) by monitoring changes in absorbance patterns, providing a rapid assessment of drug stability and shelf-life [64].
Advanced Spectrophotometric Protocols for Resolving Mixtures
When faced with overlapping spectra from drug combinations, several sophisticated yet simple mathematical spectrophotometric methods can be employed without a separation step. The following protocols for analyzing a Terbinafine HCl (TFH) and Ketoconazole (KTZ) combination, adapted from a 2025 study, exemplify this approach [37].
Third Derivative Spectrophotometry (D³)
This method effectively eliminates background interference and resolves overlapping spectra.
- Principle: The third derivative of the absorption spectrum is calculated, which narrows spectral bands and allows for the measurement of one drug at a wavelength where the derivative signal of the other is zero [37].
- Protocol:
- Preparation: Prepare standard stock solutions of TFH and KTZ at 1.0 mg/mL in methanol. Dilute with distilled water to create working solutions of 100 µg/mL.
- Calibration: Prepare a series of standard solutions in the concentration ranges of 0.6–12.0 µg/mL for TFH and 1.0–10.0 µg/mL for KTZ.
- Measurement: Using a UV-Vis spectrophotometer, record the third-order derivative spectra of the solutions with a Δλ of 8 nm and a scaling factor of 10.
- Quantification: Measure the derivative amplitudes at 214.7 nm for TFH and 208.6 nm for KTZ.
- Analysis: Construct a calibration curve by plotting the derivative amplitudes against the corresponding concentrations.
Ratio Difference Spectrophotometry
This method uses the difference in amplitudes at two points on the ratio spectrum for quantification.
- Principle: The absorption spectrum of the mixture is divided by the spectrum of a standard concentration of one drug (the "divisor"). The difference in the amplitudes of this ratio spectrum at two carefully selected wavelengths is directly proportional to the concentration of the other drug [37] [142].
- Protocol:
- Sample Prep: Prepare sample and standard solutions as in Section 4.1.
- Spectral Division:
- For TFH: Divide the spectra of the TFH standard series by the spectrum of a KTZ divisor (3.0 µg/mL).
- For KTZ: Divide the spectra of the KTZ standard series by the spectrum of a TFH divisor (4.0 µg/mL).
- Measurement: For the resulting ratio spectra, measure the difference in amplitudes (ΔP) between 222.7 nm and 204.3 nm for TFH, and between 209.8 nm and 233.2 nm for KTZ.
- Calibration: Plot the ΔP values against the respective drug concentrations to generate the calibration curves.
Table 2: Performance Data for Spectrophotometric Analysis of TFH and KTZ [37]
| Method | Analyte | Linear Range (µg/mL) | LOD (µg/mL) | LOQ (µg/mL) | Remarks |
|---|---|---|---|---|---|
| Third Derivative (D³) | TFH | 0.6 - 12.0 | < 0.3 | < 1.0 | Resolves at 214.7 nm |
| KTZ | 1.0 - 10.0 | < 0.3 | < 1.0 | Resolves at 208.6 nm | |
| Ratio Difference | TFH | 0.6 - 12.0 | < 0.3 | < 1.0 | Uses ΔP (222.7-204.3 nm) |
| KTZ | 1.0 - 10.0 | < 0.3 | < 1.0 | Uses ΔP (209.8-233.2 nm) |
The Scientist's Toolkit: Essential Reagent Solutions
The versatility of spectrophotometry is enhanced by using specific reagents that induce measurable color changes or enhance absorbance.
Table 3: Key Research Reagent Solutions in Spectrophotometry [64]
| Reagent Category | Function | Example Reagents | Common Pharmaceutical Application |
|---|---|---|---|
| Complexing Agents | Form stable, colored complexes with analytes to enhance sensitivity and enable quantification of poorly absorbing compounds [64]. | Ferric Chloride, Ninhydrin | Analysis of phenolic drugs (e.g., Paracetamol); analysis of peptides and amino acids [64]. |
| Oxidizing/Reducing Agents | Modify the oxidation state of the analyte, leading to a product with different absorbance properties, often in the visible range [64]. | Ceric Ammonium Sulfate, Sodium Thiosulfate | Determination of ascorbic acid and other antioxidants; analysis of iodine-based reactions [64]. |
| pH Indicators | Change color depending on the solution's pH, allowing for the analysis of acid-base equilibria of drugs [64]. | Bromocresol Green, Phenolphthalein | Assay of weak acids or base-forming drugs; ensuring correct pH in formulations [64]. |
| Diazotization Reagents | Convert primary aromatic amines into diazonium salts, which can couple to form highly colored azo compounds [64]. | Sodium Nitrite & Hydrochloric Acid | Analysis of sulfonamide antibiotics and other drugs containing primary amine groups [64]. |
Workflow and Decision Pathway
The following diagram illustrates a standard spectrophotometric assay workflow and the logical decision process for choosing spectrophotometry over UFLC.
Within a modern analytical laboratory, the power of a technique is measured not only by its peak performance but by its strategic fit. Spectrophotometry demonstrates its enduring value in applications where simplicity, cost-efficiency, and speed are paramount for the analysis of high-concentration APIs. The development of advanced, mathematically resolved methods further expands its utility to specific multi-component formulations, offering a green and sustainable alternative without sacrificing accuracy [37] [142]. For routine quality control, dissolution profiling, and stability studies, where the analytical targets are well-defined and concentrations are sufficiently high, spectrophotometry is not merely an adequate choice—it is an optimal one. The discerning scientist, therefore, leverages spectrophotometry not as a legacy technique, but as a powerful and rational first-line tool in a broad analytical arsenal that includes powerful separation technologies like UFLC.
The selection of an appropriate analytical method is a critical step in pharmaceutical development and quality control, directly impacting the accuracy, efficiency, and environmental footprint of the analysis. This guide provides a structured framework for researchers and drug development professionals to choose between sophisticated techniques like Ultra-Fast Liquid Chromatography (UFLC) and more accessible spectrophotometric methods. The decision is contextualized within a broader thesis research comparing these techniques, with the aim of balancing analytical performance with practical considerations. The framework integrates key performance metrics, experimental protocols, and sustainability assessments to support informed methodological choices in pharmaceutical analysis.
Fundamental Principles and Comparative Metrics
Core Analytical Principles
Spectrophotometric Methods are based on the measurement of light absorption by a substance at specific wavelengths. The fundamental relationship is described by the Beer-Lambert Law, which states that absorbance is directly proportional to the concentration of the analyte, its molar absorptivity, and the path length of the sample cell [64]. These methods utilize various reagents—including complexing agents, oxidizing/reducing agents, pH indicators, and diazotization reagents—to enhance detection and accuracy for pharmaceutical compounds [64].
Ultra-Fast Liquid Chromatography (UFLC), and its advanced form Ultra-High-Performance Liquid Chromatography (UHPLC), represents the evolution of traditional HPLC. These techniques use smaller particle sizes (<2 µm) and higher operating pressures (exceeding 1300 bar in modern systems) to achieve faster separations, greater resolution, and enhanced sensitivity compared to conventional HPLC [34] [30]. When coupled with mass spectrometry (MS), particularly tandem MS (MS/MS), the technique offers unparalleled selectivity and sensitivity for trace-level analysis in complex matrices [42] [34].
Performance Comparison
Table 1: Comparative Analysis of Spectrophotometric and UFLC Methods
| Parameter | Spectrophotometric Methods | UFLC/UHPLC Methods | UFLC-MS/MS Methods |
|---|---|---|---|
| Typical Linear Range | 0.1 - 70 µg/mL [132] [144] | Varies with detector | Varies with analyte and matrix |
| Detection Limit (LOD) | ~0.1 µg/mL (e.g., Nitisinone) [144] | Lower than spectrophotometry | 0.1 - 300 ng/L (in water) [42] |
| Analysis Time | Minutes (minimal preparation) | 10 minutes or less [42] | <10 minutes [42] |
| Multi-analyte Capability | Limited, requires resolution of overlapped spectra [132] [37] | Excellent | Excellent |
| Selectivity/Specificity | Moderate, susceptible to interference [42] | High | Very High (via MRM) [42] |
| Sample Throughput | High for single analytes | Very High | High to Very High |
| Operator Skill Level | Basic to Moderate | Moderate to High | High |
| Solvent Consumption | Low (e.g., 10 mL per sample [132]) | Low (due to fast gradients and narrow columns) | Low |
| Instrument Cost | Low | High | Very High |
| Greenness (AGREE Score) | High potential (e.g., 0.82 for some methods [37]) | Moderate to High | Improving with new systems [42] [30] |
Detailed Experimental Protocols
To illustrate the practical application of both technique categories, the following are standardized protocols for analyzing drug mixtures.
Protocol 1: Simultaneous Spectrophotometric Analysis of a Binary Drug Mixture
This protocol, adapted from methods for Paracetamol-Meloxicam analysis, is suitable for quantifying two drugs with overlapping spectra in a formulation [132].
1. Key Research Reagent Solutions
- Methanol (HPLC-grade): Primary solvent for dilution.
- Dimethylformamide (DMF): Used sparingly to dissolve poorly water-soluble drugs (e.g., Meloxicam).
- Standard Stock Solutions (1000 µg/mL): Prepared by dissolving precise weights of pure drug substances in methanol.
- Britton-Robinson Buffer (pH 6): For pH control in charge-transfer complex-based methods [144].
2. Equipment
- Double-beam UV-Vis spectrophotometer with 1-cm quartz cells.
- Software capable of recording zero-order spectra and calculating first-order derivative spectra.
3. Procedure
- Sample Preparation: Weigh and grind tablets. Extract active ingredients using methanol with 30-minute sonication. Filter into a volumetric flask and dilute to volume with methanol [132].
- Standard Preparation: Serially dilute standard stock solutions with methanol to create working solutions within the linear range (e.g., 3–30 µg/mL).
- Spectral Acquisition: Scan the zero-order absorption spectra of all standard and sample solutions from 200 to 400 nm against a solvent blank. Save the spectra.
- Data Processing for Drug A (e.g., Meloxicam):
- Zero-Order Method: If Drug A has a unique peak with no interference from Drug B (e.g., at 361 nm), measure the absorbance directly and plot against concentration.
- First-Order Derivative Method: Generate the first-derivative (1D) spectra. Measure the peak amplitude at a wavelength where Drug B shows zero-crossing (e.g., 342 nm). Plot this amplitude against the concentration of Drug A [132].
- Data Processing for Drug B (e.g., Paracetamol):
- First-Order Derivative Method: From the 1D spectra, measure the trough amplitude at a wavelength where Drug A shows zero-crossing (e.g., 262 nm). Plot this amplitude against the concentration of Drug B [132].
- Quantification: Use the respective calibration curves to determine the concentration of each drug in the sample solutions.
Diagram 1: Spectrophotometric analysis workflow for a binary drug mixture.
Protocol 2: UFLC-MS/MS Method for Trace Pharmaceutical Analysis
This protocol outlines a green/blue UHPLC-MS/MS method for sensitive, multi-analyte determination, such as in water or biological fluids [42].
1. Key Research Reagent Solutions
- Mobile Phase A: 0.1% Formic acid in water.
- Mobile Phase B: 0.1% Formic acid in acetonitrile or methanol.
- Mixed Standard Stock Solution (e.g., 1 mg/mL): Prepared in methanol for each analyte.
- Internal Standard Solution: A stable isotopically labeled analog of the target analytes.
- Solid-Phase Extraction (SPE) Cartridges: e.g., Oasis HLB or equivalent for sample clean-up and pre-concentration.
2. Equipment
- UHPLC system capable of operating at > 1000 bar pressure (e.g., Thermo Vanquish, Agilent 1290 Infinity III).
- Tandem mass spectrometer with electrospray ionization (ESI) source (e.g., Sciex 7500+).
- Analytical column: C18 column with sub-2 µm particles (e.g., 2.1 x 50 mm, 1.7 µm).
3. Procedure
- Sample Preparation (Solid-Phase Extraction):
- Condition the SPE cartridge with methanol and equilibrate with water.
- Load the sample (e.g., 100 mL of water).
- Wash with a mild solvent (e.g., 5% methanol in water).
- Elute analytes with a strong solvent (e.g., pure methanol). Note: The evaporation step can be omitted to enhance greenness [42].
- Reconstitute the eluate in the initial mobile phase composition.
- Chromatographic Separation:
- Column Temperature: 40 °C.
- Flow Rate: 0.4 mL/min.
- Injection Volume: 5-10 µL.
- Gradient Program: Initiate at 5% B, ramp to 95% B over 5-7 minutes, hold for 1-2 minutes, then re-equilibrate to initial conditions. Total run time: ~10 minutes [42].
- Mass Spectrometric Detection:
- Ionization Mode: ESI positive or negative, optimized per analyte.
- Data Acquisition: Multiple Reaction Monitoring (MRM).
- Source Parameters: Optimize source temperature, desolvation gas flow, and collision energy for each analyte.
- Data Analysis:
- Integrate peak areas for each analyte and internal standard.
- Use a calibration curve (internal standard method) for quantification.
- Sample Preparation (Solid-Phase Extraction):
Diagram 2: UFLC-MS/MS workflow for trace pharmaceutical analysis.
Method Selection Decision Framework
The choice between spectrophotometry and UFLC is not a matter of superiority but of fitness for purpose. The following structured decision pathway, based on key analytical questions, guides the selection process.
Decision Pathway
Diagram 3: Method selection decision pathway for pharmaceutical analysis.
Framework Application Guidelines
Choose Spectrophotometry When: The application involves a single analyte or a simple binary mixture where spectral overlap can be resolved mathematically (e.g., using derivative or ratio spectra) [132] [37]. It is ideal for routine quality control in formulated products, when capital and operational budgets are constrained, and when the required sensitivity is in the µg/mL range [64]. Its greenness credentials are often high due to low solvent consumption and energy requirements [132] [37].
Choose UFLC with UV/PDA Detection When: Analyzing more complex mixtures (3+ components) that spectrophotometry cannot resolve, or when higher throughput than traditional HPLC is needed. It provides superior selectivity for stability-indicating methods and impurity profiling [30].
Choose UFLC-MS/MS When: The analysis demands the highest possible sensitivity and specificity, such as for trace-level pharmaceutical contaminants in the environment [42], metabolites in biological matrices [34], or for unambiguous structural identification. It is the preferred technique for complex matrices and advanced applications like proteomics and multi-omics in drug research [34] [30].
The decision framework presented here underscores that modern pharmaceutical analysis requires a nuanced approach to method selection. Spectrophotometry remains a powerful, cost-effective, and environmentally friendly tool for well-defined applications, especially with advanced signal processing techniques. UFLC and UFLC-MS/MS offer unparalleled resolution, speed, and sensitivity for complex challenges. The optimal choice is ultimately dictated by a careful balance of the analytical problem, performance requirements, available resources, and the principles of Green Analytical Chemistry. This guide empowers scientists to make that critical choice with confidence, ensuring the generation of reliable, actionable data throughout the drug development lifecycle.
Conclusion
The choice between Ultra-Fast Liquid Chromatography and Spectrophotometry is not a matter of superiority but of strategic application. UFLC, particularly when hyphenated with mass spectrometry, offers unparalleled specificity, sensitivity, and the ability to deconvolute complex mixtures, making it indispensable for modern drug development, metabolomics, and trace impurity analysis. Spectrophotometry remains a vital, cost-effective tool for high-throughput quantitative analysis of single components where specificity is achievable. The future of pharmaceutical analysis lies in leveraging the strengths of both techniques, with continued advancements in column technology, instrument inertness, and data analysis software further pushing the boundaries of speed and accuracy. A firm grasp of both methods' principles, applications, and validation requirements empowers scientists to ensure drug safety, efficacy, and compliance, ultimately accelerating the delivery of new therapies.
References
- 1. Optimizing Ultra-High-Performance Liquid ... [https://www.intechopen.com/chapters/1188571]
- 2. A Simple Approach to Performance Optimization in HPLC ... [https://www.chromatographyonline.com/view/simple-approach-performance-optimization-hplc-and-its-application-ultrafast-separation-development]
- 3. Development of Ultra Fast Liquid Chromatography (UFLC) ... [https://www.scirp.org/journal/paperinformation?paperid=62846]
- 4. Beer-Lambert Law | Transmittance & Absorbance [https://www.edinst.com/resource/the-beer-lambert-law/]
- 5. Beer–Lambert law [https://en.wikipedia.org/wiki/Beer%E2%80%93Lambert_law]
- 6. Beer-Lambert's Law: Principles and Applications in Daily Life [https://www.findlight.net/blog/beer-lamberts-law-explained-applications/]
- 7. Comparison of HPLC and UV Spectrophotometric Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8653657/]
- 8. Comparison of UPLC and HPLC methods for determination ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814614018330]
- 9. The Beer-Lambert Law [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy/Electronic_Spectroscopy_Basics/The_Beer-Lambert_Law]
- 10. The Bouguer‐Beer‐Lambert Law: Shining Light on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7540309/]
- 11. Data-Driven FTIR Spectroscopy for the Discrimination of ... [https://www.mdpi.com/1420-3049/30/20/4083]
- 12. Derivation of Beer Lambert Law [https://byjus.com/physics/derivation-of-beer-lambert-law/]
- 13. Achieving Ultrafast Liquid Chromatography [https://www.pharmtech.com/view/achieving-ultrafast-liquid-chromatography]
- 14. Ultrafast HPLC: Different Approaches to Increased ... [https://www.chromatographyonline.com/view/ultrafast-hplc-different-approaches-increased-throughput]
- 15. Application of Very-High Pressure Liquid Chromatography ... [https://www.americanpharmaceuticalreview.com/Featured-Articles/36910-Application-of-Very-High-Pressure-Liquid-Chromatography-for-Developing-Fast-and-More-Efficient-Separations-within-Pharmaceutical-Development/]
- 16. LC-4000 Series HPLC and UHPLC - JASCO Inc [https://jascoinc.com/products/chromatography/hplc/]
- 17. HPLC/UHPLC Systems and Solutions [https://www.agilent.com/en/product/liquid-chromatography?srsltid=AfmBOoquNVo_D6FrBIQy3f_6Cih9_CTOSNirtTXJGxH2bbB86o9Rrdld]
- 18. Color Perception: How Spectrophotometry Can Neutralize ... [https://www.hunterlab.com/blog/color-perception-how-spectrophotometry-can-neutralize-the-effects-of-visual-discrimination/]
- 19. Ultrafast liquid chromatography-tandem mass spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8979757/]
- 20. LC vs. HPLC vs. UHPLC: Tracing the Evolution of ... [https://www.metwarebio.com/lc-vs-hplc-vs-uhplc-evolution-chromatography/]
- 21. How Chemometrics Revives the UV-Vis Spectroscopy ... [https://www.mdpi.com/2227-9040/11/1/8]
- 22. Comparative analysis and validation of analytical ... [https://www.sciencedirect.com/science/article/abs/pii/S0019452224000530]
- 23. LC/MS Applications in Drug Development [https://www.bioagilytix.com/blog/lc-ms-applications-in-drug-development/]
- 24. High-throughput high-performance liquid chromatography ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166900001762]
- 25. Interelement Corrections in Spectrochemistry [https://www.spectroscopyonline.com/view/interelement-corrections-spectrochemistry]
- 26. Matrix effect correction method based on the main spectral ... [https://www.sciencedirect.com/science/article/abs/pii/S0584854722000829]
- 27. 9.3: Interferences in Absorption Spectroscopy [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Instrumental_Analysis_(LibreTexts)/09%3A_Atomic_Absorption_and_Atomic_Fluorescence_Spectrometry/9.03%3A_Interferences_in_Absorption_Spectroscopy]
- 28. What still hinders the routine application of miniaturized ... [https://www.sciencedirect.com/science/article/pii/S2772391724000367]
- 29. LC/MS applications in drug development [https://pubmed.ncbi.nlm.nih.gov/10568041/]
- 30. New HPLC, MS, and CDS Products from 2024–2025 [https://www.chromatographyonline.com/view/new-hplc-ms-and-cds-products-from-2024-2025-a-brief-review]
- 31. Comparison of Quantitative Mass Spectrometry Platforms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6021760/]
- 32. Comparison of a Comprehensive Liquid Chromatography ... [https://pubmed.ncbi.nlm.nih.gov/40498580/]
- 33. Ultra-High-Performance Liquid Chromatography ... [https://pubmed.ncbi.nlm.nih.gov/39916635/]
- 34. Innovative applications and future perspectives of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11979114/]
- 35. Univariate and multivariate signal processing ... [https://www.nature.com/articles/s41598-025-22700-0]
- 36. Selective six spectrophotometric methods for determination of ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-025-01607-x]
- 37. Innovative spectrophotometric methods for the analysis of ... [https://www.nature.com/articles/s41598-025-25067-4]
- 38. Eco-friendly four spectrophotometric approaches for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11551209/]
- 39. Comparison of UV spectrophotometry and high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658086/]
- 40. Clinical validation of a liquid chromatography single ... [https://pubmed.ncbi.nlm.nih.gov/40776792/]
- 41. Development and validation of a 'dilute and shoot' LC–MS ... [https://www.sciencedirect.com/science/article/pii/S1570023225003046]
- 42. Development and validation of a green/blue UHPLC-MS ... [https://www.nature.com/articles/s41598-025-15614-4]
- 43. High-Throughput Metabolomics for Identification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7059176/]
- 44. Metabolic profiling and pharmacokinetic studies of Baihu-Guizhi... [https://link.springer.com/article/10.1186/s13020-022-00665-w]
- 45. Metabolome searcher: a high tool for throughput ... metabolite [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-015-0462-y]
- 46. Developing and Validating Dissolution Procedures [https://www.chromatographyonline.com/view/developing-and-validating-dissolution-procedures-0]
- 47. Choices of chromatographic methods as stability indicating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8027279/]
- 48. Development of Stability-Indicating Analytical Procedures ... [https://www.chromatographyonline.com/view/development-of-stability-indicating-analytical-procedures-by-hplc-an-overview-and-best-practices]
- 49. Comparative analysis using UPLC and UV methods for ... [https://www.sciencedirect.com/science/article/pii/S2211715624005952]
- 50. Development and Validation of a Discriminative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3909171/]
- 51. Development and validation of a stability indicating UPLC ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3745131/]
- 52. A validated stability-indicating UPLC method for ... [https://www.sciencedirect.com/science/article/pii/S1878535213004504]
- 53. [Determination of trace genotoxic impurities in nifedipine by ... [https://pubmed.ncbi.nlm.nih.gov/35243836/]
- 54. LC-MS vs LC-MS/MS: Techniques, Use Cases, and ... [https://www.creative-proteomics.com/resource/lc-ms-vs-lc-msms.htm]
- 55. Ultra Performance Liquid Chromatography (Uplc): A New ... [https://www.ijsrtjournal.com/article/Ultra-Performance-Liquid-Chromatography-Uplc-A-New-Trend-in-Analysis]
- 56. Spectrophotometric Color Measurement to Assess ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7699973/]
- 57. LC-MS based stability-indicating method for studying the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7714013/]
- 58. An Ultra-Fast Validated Green UPLC-MS/MS Approach ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11679331/]
- 59. ICH and FDA Guidelines for Analytical Method Validation [https://www.labmanager.com/ich-and-fda-guidelines-for-analytical-method-validation-34252]
- 60. Development and validation of spectrophotometric ... [https://www.sciencedirect.com/science/article/pii/S2666831924000699]
- 61. The Essence of Modern HPLC: Advantages, Limitations ... [https://www.chromatographyonline.com/view/essence-modern-hplc-advantages-limitations-fundamentals-and-opportunities]
- 62. Strain-Level Discrimination of Bacteria by Liquid ... - PMC - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC10407911/]
- 63. Ultra-high performance liquid chromatography-MS/MS ... [https://fjps.springeropen.com/articles/10.1186/s43094-019-0007-8]
- 64. Spectrophotometric Methods in Pharmaceutical Analysis [https://juniperpublishers.com/ijesnr/IJESNR.MS.ID.556391.php]
- 65. Spectrophotometry: Uses, Advantages & Applications [https://lifesciences.danaher.com/us/en/library/spectrophotometry.html]
- 66. A Friendly Complexing Agent for Spectrophotometric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8196596/]
- 67. The complexation color development—visible ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X24009640]
- 68. Determination of nitrite by simple diazotization method [https://www.sciencedirect.com/science/article/abs/pii/S0026265X02000930]
- 69. Development and Validation of Spectrophotometric and ... [https://www.ijpsnonline.com/index.php/ijpsn/article/view/1295]
- 70. Comparison of ultra-performance liquid chromatography ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967305021151]
- 71. What are the Key Challenges in Spectrophotometric ... [https://www.drawellanalytical.com/what-are-key-challenges-in-spectrophotometric-analysis/]
- 72. Innovations in Liquid Chromatography: 2025 HPLC ... [https://www.chromatographyonline.com/view/innovations-in-liquid-chromatography-2025-hplc-column-and-accessories-review]
- 73. Discrimination of polysorbate 20 by high-performance ... [https://www.sciencedirect.com/science/article/pii/S2095177923003027]
- 74. Application of LCMS in small-molecule drug development [https://www.europeanpharmaceuticalreview.com/article/43628/application-lcms-small-molecule-drug-development/]
- 75. Derivative UV-vis absorption spectra as an invigorated ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993615301357]
- 76. HPLC Pressure Problems? Here's How to Diagnose and Fix ... [https://www.mastelf.com/hplc-pressure-problems-heres-how-to-diagnose-and-fix-them/]
- 77. LC Troubleshooting Essentials: A Guide to Common ... [https://www.chromatographyonline.com/view/lc-troubleshooting-essentials-a-guide-to-common-problems-and-solutions-for-peak-tailing-ghost-peaks-and-pressure-spikes]
- 78. Expert Guide to Troubleshooting Common HPLC Issues [https://aelabgroup.com/expert-guide-to-troubleshooting-common-hplc-issues/]
- 79. Effective LC Troubleshooting: Symptom-Based Strategies ... [https://www.restek.com/articles/effective-lc-troubleshooting-symptom-based-strategies-and-solutions?srsltid=AfmBOoq7hMecbZy0BXmbvifnaTWoyW-RoLelsafGxQ3W5SkotmzdlXqo]
- 80. HPLC Troubleshooting Guide [https://www.thermofisher.com/us/en/home/industrial/chromatography/chromatography-learning-center/high-performance-liquid-chromatography-hplc-support/hplc-troubleshooting.html]
- 81. An Introduction to Peak Tailing, Fronting and Splitting in ... [https://www.acdlabs.com/blog/an-introduction-to-peak-tailing-fronting-and-splitting-in-chromatography/]
- 82. Peak Shapes and Their Measurements [https://www.chromatographyonline.com/view/peak-shapes-and-their-measurements-need-and-concept-behind-total-peak-shape-analysis-0]
- 83. Troubleshooting Basics, Part IV: Peak Shape Problems [https://www.chromatographyonline.com/view/troubleshooting-basics-part-iv-peak-shape-problems]
- 84. What is Peak Fronting? [https://www.perkinelmer.com/library/what-is-peak-fronting.html]
- 85. Peak Splitting in HPLC: Causes and Solutions [https://www.sepscience.com/peak-splitting-in-hplc-causes-and-solutions-9379]
- 86. Peak Tailing in HPLC [https://www.elementlabsolutions.com/uk/chromatography-blog/post/peak-tailing-in-hplc]
- 87. Total peak shape analysis: detection and quantitation of ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967317308610]
- 88. Abnormal Peak Shapes [https://www.shimadzu.com/an/service-support/technical-support/analysis-basics/tips/abnormalpeakshapes.html]
- 89. Ultra-performance liquid chromatography method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11924836/]
- 90. What is Stray light and how it is monitored? [https://lab-training.com/what-is-stray-light-and-how-it-is-monitored/]
- 91. Stray Light - an overview [https://www.sciencedirect.com/topics/engineering/stray-light]
- 92. Errors in Spectrophotometry and Calibration Procedures to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5293527/]
- 93. Improve UV Visible Spectrophotometer Measurement ... [https://www.metashcorp.com/How-to-Improve-the-Measurement-Accuracy-of-UV-Visible-Spectrophotometer.html]
- 94. Ion Suppression: A Major Concern in Mass Spectrometry [https://www.chromatographyonline.com/view/ion-suppression-major-concern-mass-spectrometry]
- 95. Strategies for the Detection and Elimination of Matrix ... [https://www.chromatographyonline.com/view/strategies-detection-and-elimination-matrix-effects-quantitative-lc-ms-analysis]
- 96. Overview, consequences, and strategies for overcoming ... [https://pubmed.ncbi.nlm.nih.gov/34546235/]
- 97. The effect of sample preparation and data processing [https://www.sciencedirect.com/science/article/abs/pii/S0039914024010373]
- 98. Assessment of matrix effect in quantitative LC-MS bioanalysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC11352789/]
- 99. UHPLC: Advancements and Challenges [https://lifesciences.danaher.com/us/en/products/chromatography-columns/topics/ultra-high-performance-liquid-chromatography-developments.html]
- 100. of Optimization Modifiers for Mobile LC-MS-Based... Phase Fast [https://pubmed.ncbi.nlm.nih.gov/36768308/]
- 101. Preparative Ultra : Shimadzu Scientific... Fast Liquid Chromatography [https://www.ssi.shimadzu.com/products/liquid-chromatography/preparative-hplc/ultra-fast-preparative-liquid-chromatography/index.html]
- 102. Development and Validation of a Stability-indicating UV ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3480759/]
- 103. Photodegradation Products and their Analysis in Food [https://www.heraldopenaccess.us/openaccess/photodegradation-products-and-their-analysis-in-food]
- 104. Photodegradation of steroid hormone micropollutants with ... [https://www.sciencedirect.com/science/article/pii/S0043135424019341]
- 105. Screening of seized emerging drugs by ultra-high ... [https://www.sciencedirect.com/science/article/abs/pii/S0379073814000425]
- 106. An overview of forensic drug testing methods and their ... [https://harmreductionjournal.biomedcentral.com/articles/10.1186/s12954-017-0179-5]
- 107. Why Signal-to-Noise Ratio Determines Limit of Detection [https://www.thermofisher.com/blog/analyteguru/hplc-troubleshooting-tips-why-signal-to-noise-ratio-determines/]
- 108. HPLC Diagnostic Skills Vol I – Noisy Baselines [https://www.elementlabsolutions.com/uk/chromatography-blog/post/hplc-noisy-baselines]
- 109. UPLC Instant Retention Time Shift [http://www.chromforum.org/viewtopic.php?t=22709]
- 110. Causes and solutions to baseline noise in liquid ... [https://www.linkedin.com/pulse/causes-solutions-baseline-noise-liquid-chromatography-lyon-jin-uup2c]
- 111. Why Your HPLC Baseline Drifts—And How to Stop It [https://www.sepscience.com/why-your-hplc-baseline-drifts-and-how-to-stop-it-11018]
- 112. HPLC Diagnostic Skills—Noisy Baselines [https://www.chromatographyonline.com/view/hplc-diagnostic-skills-noisy-baselines]
- 113. Retention times shift out in ACQUITY UPLC H-Class [https://support.waters.com/KB_Inst/Chromatography/WKB76868_Retention_times_shifting_out_in_ACQUITY_UPLC_H-Class]
- 114. Retention time shift for particular analytes - WKB90951 - Waters [https://support.waters.com/KB_Inst/Chromatography/WKB90951_Retention_time_shift_for_particular_analytes]
- 115. ACQUITY UPLC shows intermittent retention time shifts [https://support.waters.com/KB_Inst/Chromatography/WKB68278_ACQUITY_UPLC_Intermittent_Retention_Time_shifts]
- 116. Development and Validation of UFLC-MS/MS Analytical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10956121/]
- 117. (PDF) Validated spectrophotometric and spectrofluorimetric methods ... [https://www.academia.edu/73791625/Validated_spectrophotometric_and_spectrofluorimetric_methods_for_determination_of_chloroaluminum_phthalocyanine_in_nanocarriers]
- 118. Validation of UV-Vis spectrophotometric colorimetric ... [https://pubmed.ncbi.nlm.nih.gov/40366746/]
- 119. Separation of Chromatographic Co-Eluted Compounds by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8065729/]
- 120. Recent Advances in Two-Dimensional Liquid ... [https://www.chromatographyonline.com/view/recent-advances-two-dimensional-liquid-chromatography-pharmaceutical-and-biopharmaceutical-analysis]
- 121. Comprehensive Analysis of LC/MS Data Using Pseudocolor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4141469/]
- 122. An open-source toolkit to visualize mass spectrometer data [https://www.sciencedirect.com/science/article/pii/S2667145X21000328]
- 123. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 124. Determining LOD and LOQ Based on the Calibration Curve [https://www.sepscience.com/hplc-solutions-126-chromatographic-measurements-part-5-determining-lod-and-loq-based-on-the-calibration-curve-6959]
- 125. Comparison of UPLC and HPLC methods for determination ... [https://pubmed.ncbi.nlm.nih.gov/25577057/]
- 126. LOD on HPLC vs UPLC [http://www.chromforum.org/viewtopic.php?t=121682]
- 127. Development and comparison of UPLC-ESI-MS and RP ... [https://pubs.rsc.org/en/content/articlehtml/2021/ra/d1ra03521e]
- 128. Development and validation of a fast ultra‐high‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8456811/]
- 129. Expediting online liquid chromatography for real-time ... [https://www.sciencedirect.com/science/article/pii/S002196732400387X]
- 130. High-Performance Liquid Chromatography Market Size to ... [https://www.precedenceresearch.com/high-performance-liquid-chromatography-market]
- 131. Advancing clinical biochemistry: addressing gaps and driving ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12011881/]
- 132. Simple spectrophotometric methods for the quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560461/]
- 133. Chromatography 2025: Sample Preparation Goes Automated [https://www.sepscience.com/chromatography-2025-sample-preparation-goes-automated-11259]
- 134. ICH Q2(R2) Validation of analytical procedures [https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline]
- 135. 3 Key Regulatory Guidelines for Method Validation [https://altabrisagroup.com/regulatory-guidelines-for-method-validation/]
- 136. Liquid Chromatography‒Tandem Mass Spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12026233/]
- 137. Trace analysis in complex mixtures using a high ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708512003573]
- 138. Simultaneous qualitative and quantitative analysis to ... [https://www.sciencedirect.com/science/article/pii/S1872204024001336]
- 139. a versatile tool for quantitative trace analysis in complex ... [https://pubmed.ncbi.nlm.nih.gov/21047638/]
- 140. Recent Advances in Liquid Chromatography–Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204688/]
- 141. Spectrophotometers in the Lab: Uses & Applications [https://bostonmedsupply.com/spectrophotometers-in-the-lab-how-they-work-and-applications/]
- 142. Selective six spectrophotometric methods for determination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12372283/]
- 143. Spectrophotometry in Teaching Labs: A Comprehensive ... [https://www.denovix.com/spectrophotometry-in-teaching-labs-a-comprehensive-guide/]
- 144. Journal of Reports in Pharmaceutical Sciences [https://brieflands.com/journals/jrps/articles/160785]