GC-MS in Forensic Science: Principles, Applications, and Advanced Methodologies for Researchers
This article provides a comprehensive examination of Gas Chromatography-Mass Spectrometry (GC-MS) as a cornerstone analytical technique in forensic science.
GC-MS in Forensic Science: Principles, Applications, and Advanced Methodologies for Researchers
Abstract
This article provides a comprehensive examination of Gas Chromatography-Mass Spectrometry (GC-MS) as a cornerstone analytical technique in forensic science. Tailored for researchers, scientists, and drug development professionals, it explores the fundamental principles of GC-MS operation, from sample separation to mass spectral detection. It details cutting-edge methodological applications in analyzing seized drugs, toxicological samples, and trace evidence like fibers. The content further offers practical guidance on troubleshooting common issues, optimizing performance, and validates the technique through comparisons with advanced methods like comprehensive two-dimensional GC (GC×GC-MS), all within the critical context of legal admissibility standards such as the Daubert and Frye criteria.
The GC-MS Foundation: Core Principles and Instrumentation for Forensic Analysis
Hyphenated techniques represent a paradigm shift in analytical science, born from the powerful synergy of combining separation methodologies with advanced spectroscopic detection technologies. The term "hyphenation" was formally introduced by Hirschfeld to describe the on-line coupling of a separation technique with one or more spectroscopic detection techniques [1] [2]. This integration creates an analytical system where the whole becomes significantly greater than the sum of its parts. By marrying the separation capabilities of chromatography with the identification power of spectroscopy, hyphenated techniques provide researchers with an unparalleled ability to characterize complex mixtures in a single, automated workflow [3].
In the specific context of forensic science, where samples are often complex mixtures and evidentiary value hinges on unambiguous identification, these techniques have become indispensable. They have evolved from novel approaches to fundamental tools that now form the analytical backbone of modern forensic laboratories, particularly in the realm of controlled substance analysis [4]. The core principle is elegant in its simplicity yet profound in its impact: a chromatograph (such as a Gas Chromatograph) separates the components of a mixture in time, and a spectrometer (such as a Mass Spectrometer) then provides definitive structural information for each separated component [1] [5]. This combination effectively addresses the key limitation of chromatography alone, which struggles to identify unknown compounds, and the key limitation of spectroscopy alone, which requires pure analytes for accurate analysis [5] [6].
The Fundamentals of GC-MS
Gas Chromatography-Mass Spectrometry (GC-MS) stands as one of the most established and reliable hyphenated techniques, often regarded as a "gold standard" in forensic and analytical chemistry for the definitive identification of substances [3] [4]. Its power derives from the seamless fusion of two powerful techniques: Gas Chromatography (GC), which excels at separating volatile components of a mixture, and Mass Spectrometry (MS), which provides a unique molecular fingerprint for each separated component.
Principles of Gas Chromatography
The gas chromatograph functions as the separation engine of the system. A sample is injected into a heated inlet, where it is instantly vaporized. A steady flow of inert carrier gas (such as helium or hydrogen) transports the vaporized sample through a long, narrow column [5] [4]. Inside this column, which is coated with a thin film of stationary phase, the different components in the sample interact with the coating to varying degrees. These differential interactions cause each compound to travel through the column at a distinct speed, leading to their separation in time [5]. The time taken for a compound to exit the column is its retention time, a characteristic parameter used in qualitative analysis [1].
Principles of Mass Spectrometry
As each separated component elutes from the GC column, it enters the mass spectrometer through a heated transfer line [5]. The core of the MS involves three fundamental processes:
- Ionization: The neutral molecules are ionized, most commonly by Electron Ionization (EI). In EI, a high-energy electron beam (typically 70 eV) bombards the molecules, causing them to lose an electron and form positively charged molecular ions [5] [4].
- Mass Analysis: These molecular ions are often unstable and fragment into smaller, characteristic ions. The resulting cocktail of ions is then separated based on their mass-to-charge ratio (m/z) by a mass analyzer [5]. Several types of analyzers exist, each with its own advantages, as detailed in Table 1.
- Detection: The separated ions are detected, and their abundances are measured. The result is a mass spectrum: a plot of ion abundance versus m/z that serves as a unique fingerprint for the compound [3] [5].
The combination of a compound's GC retention time and its MS fragmentation pattern provides a powerful, two-dimensional identifier that offers a very high level of confidence in confirming the identity of an unknown substance in forensic analysis [5] [4].
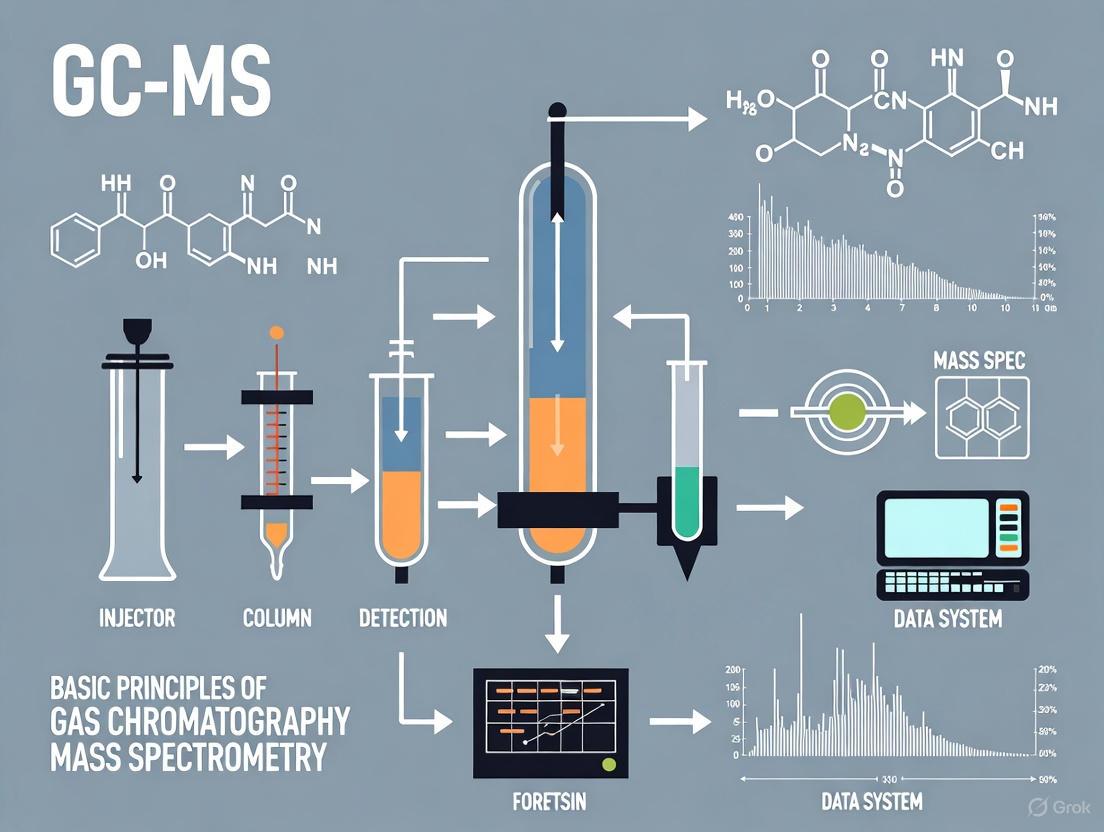
GC-MS Workflow
The following diagram illustrates the integrated workflow of a GC-MS system, from sample injection to data analysis:
GC-MS Instrumentation and Methodologies
A deep understanding of the components and operational parameters of a GC-MS system is crucial for developing robust forensic methods. The instrument is composed of two main building blocks: the gas chromatograph and the mass spectrometer, interfaced to maintain the integrity of the separation and the vacuum of the MS [4].
Key Instrumental Components
- Carrier Gas System: Requires a high-purity, chemically inert gas such as Helium, Hydrogen, or Nitrogen. The gas is regulated by a pressure controller to ensure a constant flow rate through the system, typically in the range of 1-3 mL/min for capillary columns [5] [4].
- Injection Port: The sample is introduced here, often via an auto-sampler. The port is heated to vaporize the liquid sample instantly. For capillary columns, a split/splitless injector is standard, allowing for the injection of very small volumes (e.g., 1 µL) in a split mode to prevent column overload, or in a splitless mode for trace analysis [4].
- GC Column: The heart of the separation. Modern forensic applications primarily use capillary columns (e.g., 10-30 m length, 0.1-0.25 mm internal diameter) which are coated with a thin film of stationary phase. Common stationary phases include 5% phenyl polysiloxane (e.g., HP-5) for a wide range of analytes, and polyethylene glycol (e.g., Carbowax) for more polar compounds [5] [4].
- Ion Source: This is where the neutral molecules are ionized after eluting from the GC column. Electron Ionization (EI) is the most prevalent method in forensic GC-MS because it produces extensive, reproducible fragmentation patterns that can be matched against standard reference libraries [5] [4].
- Mass Analyzer: This component separates the ions based on their m/z. The choice of analyzer depends on the application requirements, such as sensitivity, resolution, and acquisition speed, as detailed in Table 1.
- Detector and Data System: The separated ions are detected, typically by an electron multiplier, generating an electronic signal that is processed by specialized software to produce chromatograms and mass spectra [5].
Table 1: Common Mass Analyzers Used in GC-MS
| Analyzer Type | Mass Resolution | Key Principle | Common Use in Forensics |
|---|---|---|---|
| Quadrupole | Unit Mass | Filters ions using DC/RF voltages [5] | Routine targeted screening and quantification; can operate in Full Scan or Selected Ion Monitoring (SIM) mode for higher sensitivity [5]. |
| Ion Trap | Unit Mass | Traps and ejects ions sequentially based on m/z [5] | Structural elucidation through MS/MS experiments, useful for confirming unknowns. |
| Time-of-Flight (ToF) | Unit to High | Measures the time ions take to travel a fixed distance [5] | Untargeted screening and identification of unknowns due to fast acquisition and high mass accuracy. |
Experimental Protocol for Forensic Drug Screening
The following is a detailed methodology for the analysis of a suspected drug sample using GC-MS, incorporating key steps from sample preparation to data interpretation.
Sample Preparation:
- Extraction: Accurately weigh ~10 mg of the homogenized solid sample. Add 10 mL of a suitable organic solvent (e.g., Methanol, Chloroform) and vortex mix for 2 minutes to extract the analytes. For biological fluids like urine or blood, a liquid-liquid or solid-phase extraction (SPE) is required to remove matrix interferences [4].
- Derivatization (if needed): For compounds with polar functional groups (e.g., alcohols, carboxylic acids) that are not amenable to GC, a derivatization step may be necessary. This involves reacting the analyte with a reagent like MSTFA (N-Methyl-N-trimethylsilyltrifluoroacetamide) to produce a more volatile and thermally stable derivative [1].
- Filtration and Dilution: Pass the extract through a 0.45 µm PTFE syringe filter. Dilute the filtrate appropriately with solvent to bring the analyte concentration within the linear dynamic range of the instrument.
Instrumental Conditions:
- GC Column: HP-5ms (30 m length × 0.25 mm ID × 0.25 µm film thickness) or equivalent [4].
- Carrier Gas: Helium, constant flow mode at 1.0 mL/min.
- Inlet Temperature: 250°C, splitless injection mode with a 1 µL injection volume.
- Oven Temperature Program: Initial temperature 70°C (hold 2 min), ramp at 20°C/min to 300°C (hold 5 min). Total run time: 16.5 min.
- MS Conditions: Ionization mode: EI, 70 eV. Ion source temperature: 230°C. Quadrupole temperature: 150°C. Solvent delay: 3 min. Data acquisition: Full scan mode, m/z range 40-550 [5] [4].
Data Analysis and Interpretation:
- Chromatogram Examination: Review the Total Ion Chromatogram (TIC) for the presence and retention times of all peaks.
- Library Searching: For each peak of interest, extract its mass spectrum and perform a library search against a commercial database (e.g., NIST/EPA/NIH Mass Spectral Library). A high similarity index (often >85%) provides preliminary identification [5].
- Confirmation: Confirm the identity by comparing the retention time and mass spectrum of the unknown with those of a certified reference standard analyzed under identical conditions [5] [4].
Applications in Forensic Research and Drug Development
GC-MS has cemented its role as a fundamental tool in forensic science and pharmaceutical research due to its specificity, sensitivity, and robustness. Its applications span from initial drug discovery to the final presentation of evidence in a court of law.
Core Forensic and Pharmaceutical Applications
Table 2: Key Applications of GC-MS in Forensics and Drug Development
| Application Area | Specific Use-Cases | Value Provided by GC-MS |
|---|---|---|
| Forensic Toxicology | Screening and confirmation of drugs of abuse (e.g., cannabinoids, opioids, amphetaines) and their metabolites in biological fluids [3] [4]. | Provides unambiguous identification required for legal proceedings; high sensitivity allows detection of trace levels. |
| Controlled Substance Analysis | Identification of unknown powders, pills, and plant materials; determination of purity and cutting agents [4]. | Considered a "gold standard"; the mass spectrum acts as a definitive fingerprint for the drug molecule itself. |
| Arson and Fire Investigation | Identification of ignitable liquid residues (e.g., gasoline, kerosene) in fire debris [3]. | Can separate and identify complex mixtures of hydrocarbons, providing evidence of accelerant use. |
| Pharmaceutical Development | Analysis of drug metabolism and pharmacokinetics (DMPK); identification of synthetic impurities and degradation products [3]. | Enables tracking of a drug and its metabolites in biological systems, crucial for safety and efficacy studies. |
The Scientist's Toolkit: Essential Reagents and Materials
Successful GC-MS analysis relies on a suite of high-purity reagents and consumables. The following table details key items used in a typical forensic drug analysis protocol.
Table 3: Essential Research Reagent Solutions for GC-MS Analysis
| Item Name | Function / Purpose |
|---|---|
| High-Purity Solvents (e.g., Methanol, Chloroform) | To dissolve and extract the analyte from the sample matrix without introducing interfering contaminants [4]. |
| Certified Reference Standards | Pure compounds of known identity and concentration used for calibration, method validation, and confirmation of analyte identity via retention time matching [5]. |
| Derivatization Reagents (e.g., MSTFA, BSTFA) | To chemically modify polar, non-volatile analytes into volatile, thermally stable derivatives suitable for GC-MS analysis [1]. |
| Internal Standards (e.g., deuterated analogs of target analytes) | Added in a known amount to the sample; used to correct for variability in sample preparation and instrument response, improving quantitative accuracy [5]. |
| Solid-Phase Extraction (SPE) Cartridges | Used for sample clean-up and pre-concentration of analytes from complex matrices like blood or urine, reducing ion suppression and extending column life [1] [2]. |
The advent of hyphenated techniques like GC-MS has fundamentally transformed the landscape of analytical chemistry, particularly in the demanding field of forensic science. By uniting the superior separation power of gas chromatography with the unequivocal identification capabilities of mass spectrometry, this technique provides a comprehensive solution for analyzing complex mixtures. It delivers the high specificity and sensitivity required to meet the rigorous standards of evidence in legal contexts and the stringent demands of drug development pipelines. As these technologies continue to evolve, with advancements in areas like fast GC, high-resolution mass spectrometry, and sophisticated data processing software, their power to separate, identify, and quantify will only grow. This ongoing innovation ensures that GC-MS will remain an indispensable cornerstone of analytical science, enabling researchers and forensic experts to uncover the truth hidden within even the most complex of samples.
Gas Chromatography-Mass Spectrometry (GC-MS) combines two powerful analytical techniques to separate, identify, and quantify complex mixtures of chemical compounds. This hyphenated system brings together the exceptional separation power of gas chromatography with the unparalleled identification capabilities of mass spectrometry. In forensic research, this makes GC-MS an indispensable tool for detecting and confirming the presence of specific substances, from drugs and toxins to trace evidence like fiber dyes or ignitable liquids [7]. The instrument works on the fundamental principle of first separating the volatile components of a sample in the GC, then ionizing and fragmenting these molecules in the MS to produce distinctive mass spectra that serve as chemical fingerprints for identification [8] [5]. The following diagram illustrates the complete pathway of an analyte through the GC-MS system, from injection to detection.
The Gas Chromatograph (GC): Separation Module
The gas chromatograph serves as the front-end of the system, responsible for the physical separation of a sample's individual components. This is critical because it ensures that compounds enter the mass spectrometer one at a time, allowing for clean and unambiguous identification [5].
Core Components & Function
- Carrier Gas: An inert gas, such as helium, hydrogen, or nitrogen, which transports the vaporized sample through the system. The carrier gas must be chemically inert to avoid reacting with the sample or the instrument components [9] [10]. The choice of gas involves a trade-off between efficiency, safety, and cost, as summarized in Table 1.
- Inlet / Vaporization Chamber: The sample introduction point. For liquid samples, the inlet is heated to instantly vaporize the sample and mix it with the carrier gas stream [5]. Common types include split/splitless injectors for capillary columns.
- Analytical Column: The heart of the separation. This is a long, thin fused-silica capillary tube (typically 10-30 meters long and 0.1-0.25 mm in internal diameter) coated on the inside with a thin layer of stationary phase [5]. As the vaporized compounds are carried through the column by the carrier gas, they interact with the stationary phase. Differences in these interactions—based on a compound's boiling point and polarity—cause each compound to spend a different amount of time in the column, leading to separation [8]. The time a compound takes to travel through the column to the detector is its retention time, a key identifying characteristic [10].
Table 1: Comparison of Common GC Carrier Gases
| Carrier Gas | Pros | Cons |
|---|---|---|
| Hydrogen (H₂) | High diffusivity for good separation efficiency; short analysis time [9] | Flammable; not completely inert and may react with some compounds [9] |
| Helium (He) | Inert, non-flammable, and safe; provides high resolution [9] | Expensive and not always readily available [9] |
| Nitrogen (N₂) | Cheap and easily available [9] | Lower separation resolution; longer analysis time; not ideal for temperature-programmed analysis [9] |
The Mass Spectrometer (MS): Detection and Identification Module
Once separated, the neutral molecules eluting from the GC column are analyzed by the mass spectrometer. The MS acts as a highly informative detector that identifies compounds based on their mass and structure [8].
The Ion Source: Creating Charged Particles
The first component the separated chemicals encounter in the MS is the ion source. Here, neutral molecules are converted into charged ions, making them susceptible to electric and magnetic fields. The most common method is Electron Ionization (EI) [8] [5]. In an EI source, a heated metal filament emits a beam of high-energy electrons (typically 70 eV). When a molecule passes through this beam, it is bombarded, causing it to lose an electron and form a positively charged molecular ion (M⁺•). This molecular ion is often unstable and possesses excess energy, leading it to break apart into smaller, characteristic fragment ions [8] [5]. The pattern of these fragments is highly reproducible and specific to the molecular structure of the compound.
The Mass Analyzer: Sorting Ions by Mass
The fragment and molecular ions are then accelerated into the mass analyzer, which separates them based on their mass-to-charge ratio (m/z). For most ions generated by EI, the charge is +1, so m/z effectively represents the mass of the ion [5]. The most common type of mass analyzer for benchtop GC-MS is the quadrupole [8] [11]. A quadrupole consists of four parallel rods to which a combination of DC and radio frequency (RF) voltages are applied. By rapidly varying these voltages, the quadrupole can act as a mass filter, allowing only ions of a specific m/z to pass through to the detector at any given moment. The instrument rapidly scans across a predetermined mass range, building a mass spectrum for each point in time during the chromatographic run [8].
The Ion Detector: From Ions to Signal
The ions that pass through the mass analyzer strike the detector, typically an electron multiplier. This device amplifies the minute current produced by the arriving ions into a measurable electrical signal [8] [11]. The signal is then sent to a computer system, which processes it to generate the chromatograms and mass spectra used for analysis [5].
Data Analysis: From Raw Signal to Forensic Evidence
GC-MS data is three-dimensional, comprising retention time, signal intensity (abundance), and mass-to-charge ratio (m/z) [5]. This rich dataset can be interrogated in several ways to perform both qualitative and quantitative analysis.
Primary Data Acquisition Modes
- Total Ion Chromatogram (TIC): The TIC is the most fundamental chromatogram. It is constructed by summing the intensities of all ions detected at each point in time during the GC run. It provides a universal detection view, where each peak represents a different compound (or co-eluting compounds) exiting the GC column [11] [5].
- Mass Spectrum: At every second along the TIC, a full mass spectrum can be obtained. This spectrum is a "fingerprint" that plots the intensity of ions versus their m/z. This fingerprint can be searched against extensive commercial spectral libraries for identification [11] [10]. Key features of a mass spectrum include the molecular ion (the heaviest ion, representing the intact molecule) and the base peak (the most intense fragment ion) [11].
Advanced Techniques for Targeted Analysis
For more sensitive or specific analysis, especially in complex matrices common in forensics, other data processing and acquisition modes are employed, as illustrated in the workflow below.
- Extracted Ion Chromatogram (EIC): After a full scan data file is acquired, the data system can extract the signal for a specific ion or set of ions. This is a powerful software-based filtering technique that simplifies the chromatogram, reduces background noise, and helps confirm the identity of a target compound by monitoring its characteristic ions [11].
- Selected Ion Monitoring (SIM): In this acquisition mode, the mass spectrometer is programmed before the analysis to monitor only a few specific ions characteristic of the target analytes. By ignoring all other ions, the instrument can spend more time measuring the signals of interest, resulting in significantly improved sensitivity and lower detection limits for quantitative analysis [11] [5]. This is distinct from EIC, as it is a separate experiment, not a data processing technique [11].
Experimental Protocol: Forensic Fiber Analysis by GC-MS
The following detailed methodology, adapted from a 2025 study, demonstrates the application of GC-MS/MS for the forensic discrimination of polyester fibers based on their dye content [12].
Application: Discrimination of single polyester fibers for forensic comparison. Objective: To identify aromatic amines derived from azo dyes within polyester fibers, providing a chemical signature for sample differentiation.
Materials and Reagents
Table 2: Key Research Reagent Solutions for GC-MS Forensic Fiber Analysis
| Reagent / Material | Function in the Protocol |
|---|---|
| Chlorobenzene | Solvent for extraction of disperse azo dyes from the polyester fiber matrix [12]. |
| Sodium Dithionite | Reducing agent that cleaves the azo bonds (-N=N-) in the dyes to yield aromatic amines [12]. |
| Citrate Buffer | Provides a stable pH environment for the controlled reductive cleavage reaction [12]. |
| Chloroform / 1,2-Dichloroethane | Extraction solvents used in Dispersive Liquid-Liquid Microextraction (DLLME) to concentrate the aromatic amines prior to analysis [12]. |
Step-by-Step Procedure
- Sample Preparation: A single polyester fiber (~2 cm in length) is placed into a micro-vial.
- Dye Extraction: Add 100 µL of chlorobenzene to the vial and heat (e.g., 100°C for 10 minutes) to extract the disperse dyes from the fiber.
- Reductive Cleavage:
- Transfer the extract to a vial containing 1 mL of citrate buffer (pH ~6).
- Add 10 µL of a fresh sodium dithionite solution.
- Heat the mixture at 70°C for 30 minutes to reduce the azo dyes into their constituent aromatic amines.
- Analyte Concentration (DLLME):
- After reduction, cool the solution.
- Rapidly inject a mixture of 1 mL of acetonitrile (disperser solvent) and 100 µL of chloroform (extraction solvent) into the sample vial.
- Centrifuge to form a sedimented droplet of the chloroform phase, now enriched with the aromatic amines.
- Collect the droplet for analysis.
- GC-MS/MS Analysis:
- Instrument: GC system hyphenated to a triple quadrupole (tandem) mass spectrometer.
- GC Column: Standard non-polar or mid-polar capillary column (e.g., 5% diphenyl / 95% dimethyl polysiloxane).
- Ionization: Electron Ionization (EI) at 70 eV.
- Data Acquisition: Multiple Reaction Monitoring (MRM). First, analyze a reference sample in full scan mode to identify target amines and their characteristic precursor ions. Then, for high-sensitivity analysis of evidence samples, use an optimized MRM method that monitors specific precursor ion > product ion transitions for each amine.
- Data Interpretation and Discrimination: Compare the MRM chromatograms of the evidence and reference fibers. The presence or absence of specific aromatic amine signals, identified by their retention times and MRM transitions, is used to differentiate the fiber samples. The high selectivity of MRM minimizes interferences, allowing for robust discrimination even with trace-level analytes [12].
The GC-MS instrument, a synergistic combination of a separation module and an identification module, provides a powerful platform for forensic analysis. Deconstructing its components—from the GC inlet and column to the MS ion source, quadrupole analyzer, and electron multiplier—reveals the sophisticated engineering that enables the detection and definitive identification of chemical substances. The versatility in data acquisition modes, from full scan for unknown identification to SIM and MS/MS for targeted, high-sensitivity quantification, makes it adaptable to a wide range of forensic challenges. As demonstrated in the fiber analysis protocol, the sensitivity and selectivity of modern GC-MS/MS systems allow forensic scientists to extract meaningful chemical data from minute traces of evidence, solidifying its status as a cornerstone technique in forensic chemistry research and casework.
Gas chromatography (GC) is a cornerstone analytical technique in forensic science, providing the critical ability to separate, identify, and quantify volatile components within complex mixtures encountered as evidence [13]. When coupled with mass spectrometry (MS), it forms gas chromatography-mass spectrometry (GC-MS), a powerful tool that is a primary pillar in analytical chemistry for characterizing chemical substances [14]. In forensic contexts, this capability is essential for analyzing diverse materials such as ignitable liquid residues in fire debris, controlled substances in seized drugs, and other trace evidence, providing scientific data that can determine whether a fire was intentionally set or identify the composition of an unknown powder [15] [13].
The fundamental principle of GC separation lies in the differential partitioning of analytes between a stationary phase and a mobile gas phase [16]. The sample is vaporized and transported by an inert carrier gas through a column containing the stationary phase. Separation occurs because each component interacts differently with the stationary phase; compounds with stronger affinities are retarded, while those with weaker interactions elute more rapidly [13]. This process results in the physical separation of mixture components over time, allowing for their individual detection and measurement [16]. The distribution constant (Kc), which is the ratio of a compound's concentration in the stationary phase to its concentration in the mobile phase, governs this movement and is the chemical parameter that enables chromatographic separation [16].
Core Principles of Chromatographic Separation
The Separation Mechanism
At its core, the gas chromatograph functions as a highly efficient distillation system. Separation hinges on two primary physicochemical properties of the analytes: their relative volatility and their affinity for the stationary phase [13]. As the vaporized sample is carried through the column by the mobile phase (carrier gas), each analyte continually partitions between the two phases. Analytes with higher volatility and lower affinity for the stationary phase spend more time in the mobile gas phase and move through the column more quickly, resulting in shorter retention times. Conversely, analytes with lower volatility and higher stationary phase affinity are delayed, exhibiting longer retention times [16] [13]. This differential migration is the fundamental mechanism that resolves a complex mixture into its individual components as they exit the column.
Key Instrumental Components
A gas chromatograph consists of several integrated subsystems, each critical to achieving a successful separation. The sequence of components and their functions are visualized in the following workflow:
The process begins at the injection port, where a precise amount of sample is introduced, typically via a syringe or autosampler, and instantly vaporized in a heated chamber [13]. The vaporized sample is then mixed with the carrier gas—an inert gas such as helium or nitrogen—which acts as the mobile phase to transport the sample through the system [16] [13]. The heart of the instrument is the column, a long, coiled tube housed within a temperature-controlled oven. The interior of the column is coated with the stationary phase, a material that selectively interacts with the analytes [16] [13]. Modern systems primarily use capillary columns with minimal inner diameters to achieve high separation efficiency. Finally, as separated components exit the column, they enter a detector. In GC-MS, this is a mass spectrometer, which ionizes the molecules, separates the ions by their mass-to-charge ratio, and provides data for both identifying and quantifying each compound [13] [15].
Forensic Applications and Experimental Protocols
Analysis of Ignitable Liquid Residues in Fire Debris
The identification of ignitable liquid residues (ILRs) in fire debris is one of the most prevalent applications of GC-MS in forensic laboratories [15]. Its objective is to determine whether materials recovered from a fire scene contain traces of accelerants like gasoline or diesel fuel, which can indicate arson. The analysis involves a multi-step protocol designed to extract and identify volatile organic compounds from complex, burned substrate matrices.
A detailed methodology for this analysis, adapted from forensic literature, is outlined below [15]:
- Sample Preparation: Simulated fire debris samples are created by spiking a substrate (e.g., wood, carpet) with a neat ignitable liquid (e.g., gasoline, diesel fuel). The substrate is then burned to simulate real fire conditions. For analysis, volatile compounds are extracted from the debris using techniques like passive-headspace extraction, often involving heating the sample in a sealed container to drive volatiles into the headspace, where they are adsorbed onto a carbonaceous strip [15].
- Sample Introduction: The extracted residues are desorbed from the collection strip using a small volume of solvent like dichloromethane. An aliquot of this solution is then introduced into the GC-MS system. Rapid screening methods may use a short (e.g., 2 m) GC column for fast analysis (≈1 minute) [15].
- Chromatographic Separation: The sample is vaporized in the injection port at high temperatures (e.g., 250°C). Separation is achieved using a non-polar or low-polarity column, such as a 100% polydimethylsiloxane (DB-1) column. A common temperature program involves a rapid ramp to separate volatile components efficiently. For rapid GC-MS, the oven may be held isothermal at a high temperature (e.g., 280°C) to prevent recondensation of analytes [15].
- Detection and Identification: Separated components eluting from the column are detected by the mass spectrometer. Data analysis involves examining the total ion chromatogram (TIC) and utilizing extracted ion profiles (EIPs). EIPs are crucial for isolating characteristic ions of target compounds (e.g., alkylbenzenes, polyaromatic hydrocarbons) from complex background interference generated by the burned substrate. Data deconvolution software is often employed to aid in identifying co-eluting peaks [15].
Advanced Visualization: Stained Chromatograms and Substance Maps
Emerging visualization techniques enhance the interpretability of complex forensic data. One advanced method involves "staining" or color-coding gas chromatograms based on mass spectral similarity [14]. In this technique:
- Recorded mass spectra are compared and sorted according to their similarity on a self-organizing map that was pre-trained on a large mass spectral database.
- The location of a mass spectrum on this map is then converted into a specific color using the hue, saturation, and lightness (HSL) color space.
- The resulting color-coded peaks in the chromatogram immediately convey the structural similarity and likely substance class of the detected analytes, facilitating rapid identification and inspection [14].
Furthermore, these data can be converted into substance maps, which are retention-time-independent summaries of the chromatogram. These maps provide an overview of the analytes present in a sample, independent of the specific analytical conditions used for separation. They are particularly useful for directly comparing samples analyzed with different GC setups and for quantifying structurally similar compounds that elute at different times [14].
Quantitative Data and Research Reagents
Performance Data for Forensic GC-MS Analysis
The efficacy of GC-MS methods is quantified through rigorous validation. The table below summarizes key performance characteristics for a rapid GC-MS method developed for ignitable liquid analysis, demonstrating the sensitivity achievable for target compounds [15].
Table: Limits of Detection (LOD) for Compounds in Ignitable Liquids via Rapid GC-MS
| Compound Name | Chemical Category | Limit of Detection (mg/mL) |
|---|---|---|
| p-Xylene | Aromatic | 0.012 |
| n-Nonane | Alkane | 0.018 |
| 1,2,4-Trimethylbenzene | Aromatic | 0.015 |
| n-Decane | Alkane | 0.016 |
| 1,2,4,5-Tetramethylbenzene | Aromatic | 0.014 |
| 2-Methylnaphthalene | Polycyclic Aromatic Hydrocarbon | 0.017 |
| n-Tridecane | Alkane | 0.013 |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of forensic GC-MS analysis relies on a suite of specialized reagents and consumables. The following table details key materials and their functions within the analytical workflow.
Table: Essential Research Reagent Solutions and Materials for Forensic GC-MS
| Item Name | Function / Application |
|---|---|
| DB-1 / DB-5 MS GC Columns | Non-polar or low-polarity stationary phases (100% polydimethylsiloxane or 5% phenyl-equivalent) standard for separating a wide range of organic compounds encountered in forensic samples like ignitable liquids and drugs [15]. |
| Helium Carrier Gas | An inert, high-purity (99.999%) mobile phase that transports the vaporized sample through the column without reacting with analytes [15]. |
| C7-C30 n-Alkane Standard Solution | A calibrated mixture used for calculating Kovats Retention Indices, which helps identify unknown compounds by normalizing their retention times against a known scale, independent of minor instrumental fluctuations [16]. |
| Dichloromethane (DCM) | A high-purity organic solvent (≥99.9%) used to desorb volatile compounds from collection devices (e.g., carbon strips from passive-headspace extraction) and prepare standard solutions and sample extracts for injection [15]. |
| Certified Reference Materials (CRMs) | Neat, high-purity analytical standards of specific compounds (e.g., p-xylene, 2-methylnaphthalene) used for method development, calibration, and determining limits of detection [15]. |
| Mass Spectral Libraries (NIST, Wiley) | Extensive databases of known compound mass spectra. Software compares the mass spectrum of an unknown analyte from the GC-MS against these libraries to propose identifications [14]. |
Data Presentation and Accessibility in Forensic Analysis
Graphical Representation of Chromatographic Data
The primary data output from a GC or GC-MS analysis is a chromatogram—a plot of the detector's response as a function of time [17]. Each peak represents a separate analyte, with the retention time serving as a characteristic identifier and the peak area being proportional to the analyte's concentration [13]. For complex forensic samples, additional data visualization tools are essential:
- Extracted Ion Profiles (EIPs): These are chromatograms reconstructed by plotting the abundance of only one or a few selected ions. EIPs are vital for forensic applications like fire debris analysis, as they can reveal the presence of a specific class of compounds (e.g., alkanes, aromatics) that might otherwise be obscured by the complex background in the total ion chromatogram [15].
- Bar Graphs and Pareto Charts: Used in forensic reporting to compare the relative abundance of specific compounds across different samples or to show the frequency of certain evidence types [18].
- Scatter Plots: Employed in chemometrics to visualize the clustering of samples based on their chemical composition, aiding in the classification of unknown liquids or the comparison of evidence samples [17].
Adherence to Accessibility and Contrast Standards
When generating diagrams, charts, and reports for scientific publication and legal proceedings, it is imperative to ensure that all graphical elements are accessible to individuals with color vision deficiencies. This aligns with WCAG (Web Content Accessibility Guidelines) standards, which stipulate minimum contrast ratios [19] [20].
The following diagram illustrates the logical decision process for analyzing a forensic sample, incorporating high-contrast colors from the specified palette to ensure accessibility:
Key contrast guidelines for scientific diagrams include [19] [20]:
- Normal Text: A minimum contrast ratio of 4.5:1 against the background.
- Large Text (18pt+): A minimum contrast ratio of 3:1 against the background.
- Graphical Objects & UI Components: Any non-text element (e.g., diagram nodes, lines, icons) required to understand the content must have a contrast ratio of at least 3:1 against adjacent colors.
By adhering to these principles, forensic scientists ensure their findings are presented clearly, accurately, and accessibly, which is paramount when presenting complex scientific evidence in a legal context.
Gas chromatography-mass spectrometry (GC-MS) is universally recognized as the gold standard in forensic trace evidence analysis due to its unparalleled ability to separate components in complex mixtures and provide definitive identification [21]. This technique combines the superior separation power of gas chromatography (GC) with the identification capabilities of mass spectrometry (MS), creating a powerful tool for analyzing everything from ignitable liquids and drugs to sexual lubricants and automotive paint [21] [10]. In forensic contexts, the mass spectrometer functions as a sophisticated detector that generates unique chemical "fingerprints" from evidence, providing both qualitative identification (what a substance is) and quantitative measurement (how much is present) to support criminal investigations [22].
The fundamental principle of GC-MS analysis involves separating volatile components in a mixture through the gas chromatograph and then detecting them in the mass spectrometer, which identifies and measures the concentration of chemicals based on their mass-to-charge ratio (m/z) [23] [10]. This provides a three-dimensional data set: retention time from the GC, signal intensity, and mass spectral information from the MS, creating a comprehensive profile of the sample's composition [5].
Core Technology: How the Mass Spectrometer Works
The mass spectrometer comprises three essential components that work in sequence to generate forensic fingerprints: the ion source, the mass analyzer, and the detector [24]. These components operate under high vacuum (approximately 10⁻⁵ to 10⁻⁸ torr) to allow ions to travel without interference from air molecules [24].
Ionization: Creating Charged Particles
The first critical step in mass spectrometry is ionization, where neutral molecules eluting from the GC column are converted into ions. The most common ionization technique in GC-MS is electron ionization (EI), where molecules are bombarded with a high-energy stream of electrons (typically 70 electron volts) emitted from a heated filament [23] [5]. This high-energy collision knocks an electron out of the molecule, creating a positively charged molecular ion (M⁺∙), which is a radical cation [25] [24]. The substantial energy transferred during this process often causes the molecular ion to become unstable and fragment into smaller ions and neutral species [5]. This fragmentation follows predictable patterns based on the molecular structure and bond energies, creating a reproducible fingerprint that can be compared across laboratories [11].
Mass Analysis: Separating Ions by m/z
After ionization, the resulting ions are accelerated and directed into the mass analyzer, which separates them based on their mass-to-charge ratio (m/z) [26]. Most ions produced in EI carry a single charge (z=1), so the m/z value is effectively equivalent to the ion's mass [24]. Several types of mass analyzers are used in forensic applications, each with different performance characteristics [26]:
- Quadrupole Analyzers: Utilize four parallel metal rods with variable electromagnetic fields to filter ions; robust and cost-effective with unit mass resolution [26] [5].
- Ion Traps: Capture and store ions in 3D space using electromagnetic fields before sequentially ejecting them to the detector; capable of multiple stages of fragmentation (MSⁿ) for structural elucidation [26].
- Time-of-Flight (ToF) Analyzers: Separate ions by measuring the time they take to travel down a flight tube; higher mass range and faster acquisition rates, suitable for comprehensive two-dimensional GC (GC×GC) applications [5].
- High-Resolution Accurate Mass (HRAM) Analyzers (Orbitrap, Magnetic Sector): Provide exceptional mass accuracy (<5 ppm) and resolution (>100,000), enabling precise determination of elemental composition [26] [5].
Detection and Data Generation
Separated ions strike the detector, typically an electron multiplier, which amplifies the signal and records the intensity for each m/z value [23] [5]. The recorded data forms a mass spectrum—a histogram plotting m/z against relative abundance [27] [24]. The most intense peak in the spectrum is designated the base peak and assigned a relative abundance of 100%, with all other peaks scaled relative to it [25] [24]. Each data point collected across the chromatographic run contains a full mass spectrum, creating a rich, three-dimensional data set (retention time, intensity, and m/z) for forensic interpretation [5].
Forensic Applications: Chemical Fingerprints in Evidence
GC-MS analysis generates distinctive chemical fingerprints from various types of forensic evidence, providing crucial links between crime scenes, victims, and suspects.
Sexual Assault Investigations: Lubricant Analysis
In sexual assault cases where perpetrators use condoms to avoid DNA transfer, the analysis of sexual lubricants becomes critical [21]. A National Institute of Justice study revealed that approximately 30% of sexual assault kits lack probative DNA profiles, making lubricant analysis an essential alternative investigative tool [21]. Natural oil-based lubricants containing ingredients like cocoa butter, shea butter, and vitamin E present highly complex mixtures that challenge conventional GC-MS due to coelution [21]. Comprehensive two-dimensional GC-MS (GC×GC–MS) has demonstrated superior capability in these analyses, resolving over 25 different components in lubricants where traditional GC-MS showed substantial coelution between 7-20 minute retention times [21]. The resulting two-dimensional chromatographic fingerprint reveals both major and minor components, including isoparaffinic compounds and aldehydes that characterize specific lubricant formulations [21].
Automotive Evidence: Paint and Tire Rubber Analysis
Automotive paint represents chemically complex evidence frequently encountered in hit-and-run accidents and vehicle-related crimes [21]. The analysis typically focuses on the clear coat layer, which contains hindered amine light stabilizers and UV absorbers to protect underlying layers [21]. While pyrolysis-GC-MS (py-GC-MS) currently offers the highest discrimination power for automotive paints, coelution of compounds like toluene and 1,2-propandial limits differentiation of similar clear coats [21]. Py-GC×GC–MS successfully overcomes these limitations by separating coeluting compounds such as α-methylstyrene and n-butyl methacrylate, which would be unresolved in traditional GC-MS [21]. Similarly, tire rubber analysis—valuable for reconstructing vehicle trajectories in hit-and-run incidents—benefits from the enhanced separation of GC×GC–MS when analyzing complex mixtures containing over 200 components, including natural/synthetic rubber, oils, plasticizers, antioxidants, and vulcanizing agents [21].
Toxicology and Controlled Substances
Mass spectrometry applications have permeated all fields of toxicology—environmental, clinical, and forensic—for the analysis of drugs, poisons, and their metabolites [26]. GC-MS and LC-MS (liquid chromatography-mass spectrometry) provide the selectivity and sensitivity required to identify and quantify toxic substances in complex biological matrices [26]. In forensic toxicology, these techniques enable the confirmation of suspected poisons and provide prognostic information to guide patient management and legal proceedings [26]. The identification relies on both the retention time matching and the unique mass spectral fingerprint of each compound, with modern instruments capable of detecting compounds at trace levels (parts-per-billion or even parts-per-trillion) in blood, urine, and tissue samples [26].
Experimental Protocols: Forensic Methodologies
Sample Preparation for Forensic Lubricant Analysis
- Sample Collection: Swabs from evidence are collected using clean cotton swabs moistened with hexane.
- Solvent Extraction: Place swabs in glass vials with 2 mL hexane and agitate via vortex mixing for 60 seconds.
- Concentration: Transfer extract to concentrator tube and evaporate under gentle nitrogen stream to approximately 100 µL.
- Instrumental Analysis: Inject 1 µL of concentrated extract into GC-MS system using splitless injection mode at 250°C.
- Chromatographic Conditions:
- Column: 30 m × 0.25 mm ID, 0.25 µm film thickness 5% phenyl polysilphenylene-siloxane
- Oven Program: 50°C (hold 2 min), ramp to 300°C at 10°C/min (hold 10 min)
- Carrier Gas: Helium, constant flow 1.2 mL/min
- Mass Spectrometric Conditions:
- Ionization: Electron Ionization (70 eV)
- Ion Source Temperature: 230°C
- Transfer Line: 280°C
- Data Acquisition: Full scan mode (m/z 40-550) [21]
Pyrolysis-GC×GC–MS for Automotive Paint Clear Coats
- Sample Preparation: Remove ~50 µg sample from clear coat layer using sterile scalpel.
- Pyrolysis Conditions:
- Pyroprobe: CDS Analytical Pyroprobe 4000
- Temperature Program: 50°C for 2 s, ramp to 750°C at 50°C/s, hold for 2 s
- GC×GC Conditions:
- Primary Column: 30 m × 0.25 mm ID, 0.25 µm film thickness non-polar phase
- Secondary Column: 2 m × 0.15 mm ID, 0.25 µm film thickness mid-polarity phase
- Modulator: Differential flow modulation (DFM) with 2.5 s modulation period
- Oven Program: 40°C (hold 2 min), ramp to 300°C at 3°C/min
- Mass Spectrometric Conditions:
- Ionization: Electron Ionization (70 eV)
- Mass Analyzer: Quadrupole or Time-of-Flight
- Acquisition Rate: 100-200 Hz for ToF (essential for GC×GC peak capture)
- Mass Range: m/z 45-600 [21]
Data Interpretation: Reading Chemical Fingerprints
Mass Spectral Interpretation Fundamentals
A mass spectrum presents as a vertical bar graph with m/z on the x-axis and relative abundance on the y-axis [24]. Key features for interpretation include:
- Molecular Ion (M⁺∙): The heaviest ion (under normal conditions) representing the intact molecule with one electron removed; provides molecular weight information [25] [24].
- Base Peak: The most intense peak in the spectrum, assigned 100% relative abundance [25] [24].
- Fragment Ions: Lower mass ions formed by decomposition of the molecular ion; reveal structural information about the original molecule [25] [5].
- Isotope Patterns: Characteristic clusters of peaks caused by naturally occurring heavy isotopes (e.g., ¹³C, ³⁷Cl, ⁸¹Br); provide information about elemental composition [24].
The "nitrogen rule" states that organic compounds containing only carbon, hydrogen, and oxygen have an even-numbered molecular weight, while those with an odd number of nitrogen atoms have an odd-numbered molecular weight [11]. This provides valuable structural information during interpretation.
Data Acquisition Modes in Forensic Analysis
Table 1: GC-MS Data Acquisition Modes for Forensic Analysis
| Acquisition Mode | Principle | Advantages | Forensic Applications |
|---|---|---|---|
| Full Scan | Continuously records all ions across a specified mass range [10] [11] | Identification of unknowns; Library search capability; Wide analytical scope [5] [11] | Drug screening; Arson analysis (ignitable liquids); Explosives residue; Untargeted metabolomics [10] [26] |
| Selected Ion Monitoring (SIM) | Monitors only pre-selected ions characteristic of target compounds [10] [5] | Increased sensitivity (5-10x over full scan); Reduced chemical noise; Improved detection limits [5] [11] | Targeted drug confirmation; Pesticide analysis in food; Trace environmental contaminants; Quantitative analysis [10] [11] |
| Tandem Mass Spectrometry (MS/MS) | Selects precursor ion, fragments it, and monitors product ions [10] [26] | Enhanced selectivity; Reduced matrix interference; Structural elucidation [10] [26] | Complex matrix analysis; Confirmatory testing; Isotope ratio analysis [10] [5] |
Advanced GC×GC–MS Data Analysis
Comprehensive two-dimensional GC (GC×GC–MS) generates structured chromatographic fingerprints where chemically related compounds form ordered patterns in the 2D separation space [21]. For example, in lubricant analysis, isoparaffinic compounds occupy specific regions (lower arc of the chromatographic plane) while aldehydes appear in distinct locations (circled regions), creating characteristic patterns that differentiate similar formulations [21]. This structured separation allows forensic scientists to quickly identify chemical classes and recognize minor components that would be obscured by coelution in conventional GC-MS [21].
Technical Specifications and Performance Metrics
Table 2: Mass Analyzer Performance Characteristics in Forensic Applications
| Mass Analyzer Type | Mass Resolution | Mass Accuracy (ppm) | Forensic Strengths | Typical Applications |
|---|---|---|---|---|
| Quadrupole | Unit mass (~1000) [26] | 100-500 ppm [26] | Robust; Cost-effective; Good sensitivity in SIM mode [26] [5] | Routine drug analysis; Environmental contaminants; Targeted quantitation [10] [26] |
| Ion Trap (QIT) | 1,000-10,000 [26] | >50 ppm [26] | MSⁿ capability; Good full-scan sensitivity; Compact design [26] | Structural elucidation; Fragmentation studies; Volatile organic compounds [26] |
| Time-of-Flight (ToF) | 1,000-40,000 [26] | <5 ppm [26] | Fast acquisition; High mass range; Untargeted analysis capability [5] | Comprehensive screening; GC×GC applications; Metabolomics; Unknown identification [21] [5] |
| Orbitrap | Up to 150,000 [26] | 2-5 ppm [26] | Ultra-high resolution; Accurate mass measurement; Multi-analyte capability [26] | Elemental composition determination; Complex matrix analysis; Research applications [26] |
| Magnetic Sector | Up to 100,000 [26] | <1 ppm [26] | Highest precision; Isotope ratio measurements [5] | Forensic isotope ratio mass spectrometry; Doping control; Regulatory analysis [5] |
Essential Research Reagent Solutions
Table 3: Essential Reagents and Materials for Forensic GC-MS Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Chromatographic Solvents (Hexane, Methanol, Dichloromethane) | Sample extraction and preparation [21] | High purity "GC-MS grade" to reduce background interference; Hexane used for lubricant extraction [21] |
| Derivatization Reagents (MSTFA, BSTFA, PFBHA) | Enhance volatility and stability of polar compounds | Used for drugs, metabolites, and explosives residues; Reduces polarity and improves chromatographic behavior |
| Internal Standards (Deuterated analogs: THC-D₃, Cocaine-D₃, PAH-D₁₂) | Quantitation reference and quality control [22] | Correct for variability in extraction and ionization; Must be added before sample preparation [22] |
| Calibration Standards (Certified reference materials) | Instrument calibration and method validation | Traceable to national standards; Multiple concentration levels for calibration curves [22] |
| GC Columns (5% phenyl polysilphenylene-siloxane, polyethylene glycol) | Compound separation [21] [10] | Low-bleed columns recommended; Dimensions: 10-30 m × 0.1-0.25 mm ID × 0.1-0.25 µm film [21] [10] |
| Quality Control Materials (Certified quality control samples) | Method performance verification | Monitor accuracy, precision, and recovery; Essential for forensic defensibility |
Gas Chromatography-Mass Spectrometry (GC-MS) combines two powerful analytical techniques to separate, identify, and quantify volatile chemical compounds within a complex mixture. This instrumentation is a cornerstone of modern analytical laboratories, with GC-MS widely regarded as the "gold standard" for confirmatory testing in fields such as forensic toxicology and pharmaceutical quality control due to its high specificity and sensitivity [28]. The technique is particularly valued for its ability to provide both qualitative (identity) and quantitative (amount) information about sample components, even when they are present at trace levels [10].
The process begins with the gas chromatograph (GC), which separates the volatile components of a mixture based on their physicochemical properties. The separated compounds are then introduced into the mass spectrometer (MS), which acts as a detector that identifies and measures them by creating and sorting ions by their mass-to-charge ratios [29]. The data generated from this process provides a multi-dimensional profile of a sample, with retention time from the GC and mass spectral data from the MS serving as the two primary axes for compound identification and characterization. This guide will detail the interpretation of the three fundamental data components: the retention time, the mass spectrum, and the total ion chromatogram.
The Total Ion Chromatogram (TIC) is one of the most fundamental and primary data outputs from a GC-MS analysis in full scan mode. It represents the summed intensity of all mass spectral peaks belonging to the same mass scan, plotted over the entire duration of the analytical run [30] [11]. In essence, the data system constructs the TIC by adding up the signals for all ions reaching the detector at every point in time [11].
The TIC provides a universal detection profile, as every vapor-phase molecule that elutes from the GC column and reaches the mass spectrometer detector contributes to the signal [11]. This makes it an powerful tool for initial sample assessment. However, this universality also presents a challenge: the TIC includes signals from all sources, including background noise, air, water, column bleed, and other contaminants present in the carrier gas or sample matrix [11]. This can sometimes lead to an elevated baseline in the chromatogram.
Key Characteristics of a TIC
- X-Axis (Retention Time): Shows the time (in minutes) from when the sample was injected to when a compound is detected. Each peak corresponds to a separated component (or co-eluting components) reaching the detector [31].
- Y-Axis (Intensity): Represents the total abundance of all ions detected at that point in time. The area under a peak is generally proportional to the quantity of the compound present, though the response can vary between different compound types [31].
- Peak Shape and Quality: A well-functioning GC-MS system will produce sharp, symmetrical peaks. A limited scan rate in full-scan mode can sometimes result in peaks that are not perfectly Gaussian [11]. Peak width is typically very narrow, often around 0.1 minutes in temperature-programmed capillary GC [11].
Table 1: Comparison of Common GC-MS Chromatogram Types
| Chromatogram Type | Description | Primary Use | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Total Ion Chromatogram (TIC) | Sum of intensities of all ions in each mass scan [30] [11]. | General, untargeted analysis; initial sample overview. | Universal detection; good for discovering unknown components. | Can be noisy; lower sensitivity for specific targets. |
| Extracted Ion Chromatogram (EIC/XIC) | A chromatogram plotted using the signal of only a single, user-specified ion mass (m/z) extracted from the full TIC data [11]. | Confirmatory analysis; reducing background interference for a known compound. | Reduces chemical noise; improves selectivity and confidence in peak identity. | Requires prior knowledge of the target ion; not all compounds are detected. |
| Selected Ion Monitoring (SIM) | A separate experiment where the MS is programmed to detect only a pre-selected set of ions, rather than scanning a full mass range [11]. | Highly sensitive targeted quantitative analysis. | Significantly reduces noise, increasing signal-to-noise ratio and sensitivity. | Not suitable for untargeted discovery; requires precise method development. |
Retention Time: The First Dimension of Separation
Retention time (RT) is a critical parameter in GC-MS, defined as the time taken for a specific analyte to pass from the injector through the column and to reach the detector [31] [32]. It is the primary metric provided by the gas chromatography step and serves as the first key to unlocking a compound's identity.
The fundamental principle governing retention time is the distribution coefficient (K), which is the ratio of a compound's concentration in the stationary phase (the column coating) to its concentration in the mobile phase (the carrier gas) [32]. A compound with a larger distribution coefficient is retained more strongly by the stationary phase and will, therefore, have a longer retention time [32]. While a rough rule of thumb is that compounds with lower boiling points elute faster, the polarity and chemical interactions with the stationary phase are equally, if not more, important.
Factors Influencing Retention Time
Retention time is not an absolute physical constant and depends on many operational and instrumental factors [32]. When comparing data, it is critical that the analyses are performed under identical conditions.
Table 2: Key Factors Affecting GC-MS Retention Time
| Factor Category | Specific Examples | Impact on Retention Time |
|---|---|---|
| Analysis Conditions | Carrier gas flow rate, oven temperature program (rate, hold times), injection port temperature [31]. | Higher flows and temperatures generally decrease retention times. |
| Column Parameters | Column type (stationary phase chemistry), column dimensions (length, diameter, film thickness), column degradation over time [32]. | A longer column or a thicker film increases retention times. Cutting a column shortens RT [32]. |
| System Condition | Presence of active sites due to contamination, degradation of the column stationary phase (column bleed) [32]. | Can cause peak tailing, adsorption, or shifts in retention time. |
Qualitative Analysis Using Retention Parameters
For qualitative identification, the retention time of an unknown compound's peak is compared to the retention time of a known standard analyzed under the exact same GC-MS conditions [32]. A match in retention time provides supporting evidence for the compound's identity.
Because absolute retention times can be unstable, analysts often use more robust relative parameters:
- Relative Retention: The ratio of the adjusted retention time of the target compound to the adjusted retention time of a reference standard. This cancels out some effects of parameters like column length and carrier gas flow [32].
- Retention Index (RI) / Linear Retention Index (LRI): A system where the retention of an unknown compound is expressed relative to a homologous series of n-alkanes analyzed under the same conditions. LRI is particularly useful because it is simple to calculate from a temperature-programmed analysis and does not require estimating the gas hold-up time [32]. Using LRI improves the accuracy of library searches and facilitates method transfer.
The Mass Spectrum: The Second Dimension of Identification
While the GC provides retention time, the mass spectrometer adds the powerful second dimension of mass information [29]. A mass spectrum is a plot of the intensity of ion signals (relative abundance) versus their mass-to-charge ratio (m/z) [11]. It acts as a unique molecular "fingerprint" that is used for definitive identification.
The process of creating a mass spectrum begins in the ion source. A common method is electron ionization (EI), where vaporized molecules eluting from the GC column are bombarded with a stream of electrons. This high-energy collision causes the neutral molecules to lose an electron and form positively charged molecular ions, which often have excess energy and fragment into smaller, characteristic ions [29]. The resulting pattern of fragments is highly specific to the structure of the original molecule.
Key Features of a Mass Spectrum
- Molecular Ion (M⁺⁺): The ion representing the intact molecule after the loss of a single electron (M⁺⁺). It typically appears at the highest m/z value in the spectrum and its mass indicates the molecular weight of the compound [11] [33]. The "nitrogen rule" states that organic compounds with an even-numbered molecular mass contain zero or an even number of nitrogen atoms, while those with an odd-numbered mass contain an odd number of nitrogen atoms [11].
- Base Peak: The most intense peak in the mass spectrum. Its abundance is set to 100% relative abundance, and all other peaks are scaled relative to it. The base peak represents the most stable or readily formed fragment ion [11] [33].
- Fragment Ions: Peaks resulting from the breakage of chemical bonds in the molecular ion. The pattern of these fragments reveals structural information about the molecule, such as the presence of specific functional groups or side chains [29] [33].
- Isotope Peaks: Most elements have naturally occurring isotopes (e.g., ¹³C, ²H). This results in small M+1, M+2, etc., peaks accompanying the main fragment peaks. The pattern and abundance of these isotope clusters can be used to deduce the elemental composition of the fragment [11].
Diagram 1: The journey of a molecule through the mass spectrometer to generate a mass spectrum.
Integrated Data Interpretation in a Forensic Context
In forensic research, the conclusive identification of a substance is paramount. GC-MS achieves this by combining the evidence from retention time and the mass spectrum. A compound is positively identified when its identity is confirmed by both retention time matching with a known standard and mass spectral matching via library search and interpretation [11].
Experimental Protocol for Confirmatory Drug Analysis
The following detailed methodology outlines a standard protocol for the confirmatory identification of an unknown compound in a forensic drug sample.
1. Sample Preparation:
- The suspected drug sample (e.g., a powder, pill, or biological extract) is dissolved in a suitable volatile solvent such as methanol or acetonitrile.
- For complex matrices like urine or blood, additional steps like liquid-liquid extraction or solid-phase extraction are required to isolate the target analytes and remove interfering compounds [28] [10].
- A known concentration of an internal standard (a compound not expected to be in the sample) is often added to correct for variations in injection volume and instrument response.
2. Instrumental Analysis:
- GC Conditions: A small volume (e.g., 1 µL) of the prepared sample is injected into the GC inlet, which is heated (200-300°C) to instantly vaporize the sample [32]. The vapor is carried by an inert gas (e.g., Helium or Hydrogen) through a capillary column. A standard initial oven temperature program might be: hold at 60°C for 1 minute, ramp at 20°C/min to 300°C, then hold for 5 minutes. The specific column (e.g., 30m x 0.25mm, 0.25µm film, 5% phenyl polysiloxane stationary phase) is selected for its ability to separate compounds of interest.
- MS Conditions: The MS is operated in full scan mode (e.g., m/z 40-500) to collect a complete mass spectrum for every eluting compound. The ion source (EI) is typically set to 70 eV, a standard energy that produces reproducible fragmentation patterns [29]. The transfer line temperature is maintained to prevent condensation of the analytes.
3. Data Analysis and Interpretation:
- The TIC is first examined to locate all eluting peaks.
- The retention time of the unknown peak of interest is compared to the retention time of a certified reference standard of the suspected drug (e.g., cocaine), analyzed using the identical method.
- The mass spectrum at the apex of the unknown peak is extracted. This spectrum is then searched against a commercial mass spectral library (e.g., NIST/EPA/NIH Mass Spectral Library). A high similarity match (e.g., >90% with a clean background) provides strong evidence for the identity of the unknown.
- For absolute confirmation, the sample is re-analyzed using a targeted method like Selected Ion Monitoring (SIM). The MS is programmed to monitor 3-4 characteristic ions (e.g., the base peak and two other significant fragments) from the known mass spectrum of the target drug at its specific retention time. The presence of all diagnostic ions with the correct abundance ratios confirms the identity [11].
Diagram 2: A forensic workflow for the confirmatory identification of an unknown compound using GC-MS.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key consumables and reagents essential for conducting reliable GC-MS analysis in a research or forensic setting.
Table 3: Essential Reagents and Materials for GC-MS Analysis
| Item | Function / Purpose | Technical Considerations |
|---|---|---|
| Certified Reference Standards | Pure compounds of known identity and concentration used for calibration, retention time matching, and mass spectral verification. | Essential for positive identification and accurate quantification. Must be traceable to a certified source. |
| Internal Standards (IS) | A compound(s) added to all samples, calibrators, and quality controls in a known amount. | Corrects for instrument variability and sample preparation losses. Should be structurally similar but chromatographically distinct from the analytes. |
| GC-MS Grade Solvents | High-purity solvents (e.g., methanol, acetonitrile, hexane) for sample preparation and dilution. | Minimal background interference (low MS signal) prevents contamination and artifact peaks. |
| Derivatization Reagents | Chemicals (e.g., MSTFA, BSTFA) that modify polar functional groups (-OH, -NH₂, -COOH) in a sample. | Increases volatility and thermal stability of analytes, improving chromatography and detection sensitivity. |
| Inert Carrier Gases | High-purity gases (e.g., Helium, Hydrogen) used as the mobile phase to carry the sample through the GC column. | Purity (≥99.999%) is critical to prevent oxygen and moisture from degrading the column and causing baseline noise. |
| Capillary GC Columns | Fused silica tubes coated with a thin layer of stationary phase where chemical separation occurs. | Selection (phase chemistry, dimensions) is critical for resolving the target analytes from each other and from matrix interferences. |
The power of GC-MS in forensic research lies in its orthogonal approach to identification. Retention time offers a physicochemical identifier based on a compound's interaction with the chromatographic system, while the mass spectrum provides a structural fingerprint based on its fragmentation pattern. The Total Ion Chromatogram serves as the comprehensive map from which this information is extracted. By rigorously applying the principles of interpreting these three data dimensions—ensuring retention time matches with certified standards, confirming mass spectral fits with library databases, and utilizing diagnostic ion ratios—researchers and forensic scientists can achieve the high level of certainty required for definitive reporting. This multi-parameter confirmation is precisely why GC-MS remains the undisputed gold standard for confirmatory analysis in laboratories worldwide.
Gas chromatography-mass spectrometry (GC-MS) combines the separation power of gas chromatography with the identification capabilities of mass spectrometry, creating one of the most powerful analytical tools in forensic science. This technique provides a definitive method for separating complex mixtures and positively identifying individual components through their unique mass fragmentation patterns. Since its commercialization in the late 1960s, GC-MS has become established as a gold standard in forensic laboratories worldwide, providing exceptionally reliable evidence for court proceedings [34] [35]. The technique's unparalleled specificity comes from its dual separation and identification mechanism—compounds are first separated by their physicochemical interactions in the GC column, then ionized and fragmented in the mass spectrometer, generating characteristic spectra that serve as molecular fingerprints [36] [34].
The fundamental strength of GC-MS lies in its ability to provide a 100% specific test that can positively confirm the presence of a particular substance, unlike nonspecific tests that merely indicate the presence of any of several substances in a category [34]. This specificity is crucial in forensic contexts where evidentiary reliability can determine legal outcomes. As Glen P. Jackson, a Distinguished Professor of Forensic and Investigative Science, notes, GC-MS remains "by far the most trusted and commonly used instrumental method of analysis for seized drugs and ignitable liquid residues" in forensic laboratories today [35]. The technique's applications span an enormous range of evidence types, from drugs and toxicological specimens to trace evidence from crime scenes and arson investigations.
Key Evidence Types Analyzed by GC-MS
Forensic laboratories routinely employ GC-MS to analyze diverse evidence types, each presenting unique analytical challenges and requirements. The following table summarizes the primary categories of evidence examined using this technique:
Table 1: Key Evidence Types Analyzed by GC-MS in Forensic Investigations
| Evidence Category | Specific Types of Evidence | Common Analytes Detected | Forensic Significance |
|---|---|---|---|
| Drugs & Toxicology | Seized drugs, blood, urine, oral fluid, hair, tissues | Opioids, stimulants, cannabinoids, benzodiazepines, novel psychoactive substances | Determines intoxication, cause of death, drug possession, compliance monitoring |
| Arson & Fire Debris | Fire debris, accelerant residues, burned materials | Gasoline, kerosene, diesel, turpentine, other ignitable liquids | Establishes arson through accelerant identification, determines fire cause |
| Trace & Biological Evidence | Fibers, paints, polymers, fingerprints, adhesives | Synthetic polymers, dyes, plasticizers, lipid fractions, additives | Links suspects to crime scenes, authenticates materials, provides investigative leads |
| Explosives & Firearms | Explosive residues, propellants, post-blast debris | Nitroglycerin, TNT, RDX, PETN, organic gunshot residues | Identifies explosive materials, links suspects to bomb-making or shooting incidents |
| Environmental & Miscellaneous | Soil, water, air samples, food contaminants, unknown substances | Pesticides, PCBs, volatile organic compounds (VOCs), persistent organic pollutants | Identifies environmental pollutants, determines contamination sources |
Drugs and Toxicological Substances
Drug analysis represents the most frequent application of GC-MS in forensic laboratories [37]. The technique provides unambiguous identification of controlled substances in both seized drug materials and biological specimens. Recent research demonstrates its continued evolution, with methods becoming faster and more sensitive. For instance, a 2025 study developed a rapid GC-MS method that reduced analysis time from 30 to 10 minutes while maintaining excellent sensitivity for substances including cocaine, heroin, synthetic cannabinoids, and pharmaceuticals [38]. This acceleration addresses critical backlog issues in forensic laboratories while delivering reproducible results with relative standard deviations (RSDs) less than 0.25% for stable compounds [38].
In toxicological analysis, GC-MS enables the detection and quantification of drugs and their metabolites in biological matrices at trace concentrations. A novel GC-MS/MS method published in 2025 achieved remarkable sensitivity for opioids and fentanyl analogues in oral fluid, with limits of detection (LODs) ranging from 0.10 to 0.20 ng/mL [39]. This exceptional sensitivity allows forensic toxicologists to identify recent drug use from minimal sample volumes, making the technique invaluable for driving under the influence investigations and postmortem toxicology.
Arson and Fire Debris Evidence
GC-MS plays a critical role in arson investigations by identifying ignitable liquid residues (ILRs) in fire debris. The technique can detect characteristic patterns of hydrocarbons and other compounds that indicate the presence of accelerants such as gasoline, kerosene, or diesel fuel, even after extensive burning [40] [35]. The analysis involves careful sample collection from fire scenes, followed by extraction of volatile compounds using techniques like headspace concentration or purge-and-trap methods, which introduce samples into the GC-MS system [34].
The evidentiary value lies in distinguishing petroleum-based accelerants from pyrolysis products generated by burning building materials, furnishings, and other substrates. GC-MS can separate and identify complex mixtures of aromatic compounds, alkanes, and other markers specific to different classes of ignitable liquids, providing scientific evidence of arson that is routinely admitted in court proceedings [35].
Trace and Biological Evidence
The application of GC-MS to trace evidence has expanded significantly with technological advancements. Comprehensive two-dimensional gas chromatography coupled with time-of-flight mass spectrometry (GC×GC–TOF-MS) provides unprecedented resolution for complex mixtures encountered in trace evidence [41]. For example, research by Vozka and colleagues employs this approach to analyze the chemical aging of latent fingerprints, monitoring time-dependent changes in lipid composition and oxidative degradation products that can help estimate when fingerprints were deposited [41].
Similarly, GC×GC–TOF-MS is being used to profile volatile organic compounds (VOCs) released during human decomposition, creating a chemical timeline that can assist in determining time since death and improving the effectiveness of human remains detection canines [41]. In polymer analysis, GC-MS identifies specific additives, plasticizers, and stabilizers that can link materials recovered from suspects to those at crime scenes [35].
Experimental Protocols and Methodologies
Standard Workflow for GC-MS Analysis
The forensic application of GC-MS follows a systematic workflow encompassing sample collection, preparation, instrumental analysis, and data interpretation. The fundamental steps are visualized in the following diagram:
Diagram 1: GC-MS Forensic Analysis Workflow
Sample Preparation Techniques
Proper sample preparation is critical for successful GC-MS analysis in forensic applications. The choice of technique depends on the sample matrix and target analytes:
Solid-Phase Extraction (SPE): Widely used for biological fluids like urine, blood, and oral fluid. SPE cartridges with various stationary phases (C18, mixed-mode, ion-exchange) selectively retain analytes of interest while removing interfering matrix components [39] [42]. For example, a 2025 method for opioids and fentanoids in oral fluid employed SPE with recovery rates consistently exceeding 57% [39].
Liquid-Liquid Extraction (LLE): Commonly applied to seized drugs and solid samples. In the rapid GC-MS method for seized drugs, solid samples were ground and extracted with methanol using sonication, while trace samples were collected with methanol-moistened swabs followed by vortexing [38].
Headspace Sampling: Particularly valuable for volatile compounds in arson evidence and toxicological alcohols. The sample is heated in a sealed vial, and the vapor phase is injected into the GC-MS [42].
Solid-Phase Microextraction (SPME): A solvent-free technique where a coated fiber absorbs analytes from the sample headspace or liquid, then desorbs them directly in the GC injector [42].
Pyrolysis-GC-MS: Used for non-volatile polymers, paints, and fibers, where controlled thermal decomposition generates characteristic fragments that identify the original material [42] [35].
Instrumental Parameters and Method Validation
Robust GC-MS methods require careful optimization of instrumental parameters. A validated rapid screening method for seized drugs established the following conditions, which illustrate typical parameters for forensic analysis:
Table 2: Instrumental Parameters for Rapid GC-MS Drug Screening [38]
| Parameter | Configuration | Forensic Application |
|---|---|---|
| GC Column | DB-5 ms (30 m × 0.25 mm × 0.25 µm) | General drug screening, moderate polarity compounds |
| Carrier Gas | Helium, constant flow mode (2 mL/min) | Inert, efficient mobile phase |
| Injection Temperature | 280°C | Suitable for most semi-volatile drugs |
| Oven Program | Initial: 80°C (hold 0.5 min), Ramp: 45°C/min to 300°C (hold 1.5 min) | Fast separation while maintaining resolution |
| Total Run Time | 10 minutes | High-throughput casework |
| Ionization Mode | Electron Ionization (EI) at 70 eV | Reproducible, library-searchable spectra |
| Mass Analyzer | Single Quadrupole | Robust, cost-effective operation |
| Detection Mode | Full Scan (m/z 40-550) | Untargeted screening, database searchable |
Method validation follows international guidelines and assesses key parameters including selectivity, linearity, accuracy, precision, limits of detection (LOD), and limits of quantification (LOQ). For example, the validated GC-MS/MS method for opioids in oral fluid demonstrated excellent linearity (R² ≥ 0.993), precision (CV% < 15%), and impressive sensitivity with LODs of 0.10-0.20 ng/mL [39].
Advanced GC-MS Techniques in Forensic Research
High-Resolution and Tandem Mass Spectrometry
GC-MS/MS significantly enhances specificity and sensitivity by isolating precursor ions and monitoring characteristic fragmentations. The GC-MS/MS method for opioids and fentanoids used multiple reaction monitoring (MRM) to achieve unparalleled selectivity in complex oral fluid samples [39]. This approach is particularly valuable for low-concentration analytes in challenging matrices, as it effectively eliminates background interference.
High-resolution mass spectrometry (HRMS) provides exact mass measurements that enable elemental composition determination and improve confidence in compound identification. As noted in research on nontargeted screening, GC-HRMS is increasingly valuable for identifying unknown pollutants and novel psychoactive substances that may escape traditional targeted methods [43].
Comprehensive Two-Dimensional GC (GC×GC–TOF-MS)
GC×GC–TOF-MS provides dramatically increased separation power by combining two chromatographic columns with different selectivity. This technique is particularly valuable for complex forensic samples such as fingerprint residues, decomposition odors, and fire debris extracts. As Petr Vozka explains, "GC×GC–TOF-MS offers unparalleled resolution and sensitivity, allowing us to monitor subtle chemical transformations that occur as fingerprint residues age" [41]. The enhanced peak capacity enables resolution of hundreds more compounds than conventional GC-MS, providing richer chemical profiles for forensic interpretation.
Alternative Ionization Techniques
While electron ionization (EI) remains the standard for forensic GC-MS due to its reproducible spectra and extensive library compatibility, alternative ionization methods offer complementary capabilities:
Chemical Ionization (CI): A softer ionization technique that produces less fragmentation, often preserving the molecular ion for confirmation of molecular weight [34].
Atmospheric Pressure Chemical Ionization (APCI): Provides efficient ionization with minimal fragmentation, enhancing sensitivity for certain compound classes. GC-APCI has been successfully applied to pesticides, brominated flame retardants, and other environmental contaminants at ultratrace levels [43].
Cold Electron Ionization (Cold-EI): Redolecular internal energy before ionization, resulting in enhanced molecular ions while retaining structural information [34].
Essential Research Reagents and Materials
Successful GC-MS analysis requires specific reagents, reference materials, and consumables tailored to forensic applications. The following table details key components of the GC-MS research toolkit:
Table 3: Essential Research Reagents and Materials for Forensic GC-MS
| Category | Specific Items | Forensic Application |
|---|---|---|
| Reference Standards | Certified reference materials (CRMs), deuterated internal standards | Target compound identification and quantification |
| SPE Sorbents | C18, C8, mixed-mode (cation/anion exchange), polymeric phases | Selective extraction of drugs from biological matrices |
| GC Columns | DB-5ms (5% phenyl polysiloxane), polar stationary phases (wax columns) | Separation of compounds based on polarity and volatility |
| Ionization Gases | High-purity helium (carrier gas), methane (CI reagent gas) | Mobile phase and chemical ionization processes |
| Sample Collection | Headspace vials, SPME fibers, methanol-rinsed swabs | Preservation of volatile compounds and trace residues |
| Solvents | High-purity methanol, acetonitrile, hexane, derivatization reagents | Sample extraction, preparation, and analyte modification |
| Quality Control | Continuing calibration verification standards, system suitability mixtures | Ensuring instrumental performance and data reliability |
The selection of appropriate research reagents is critical for method validity and admissibility of results in legal proceedings. For example, the use of certified reference materials ensures accurate compound identification, while deuterated internal standards correct for matrix effects and recovery variations during sample preparation [39] [42].
GC-MS remains an indispensable analytical technique in modern forensic science, providing unequivocal compound identification across diverse evidence types. From its established role in drug identification and toxicology to emerging applications in fingerprint aging and odor profiling, the technique continues to evolve through advancements in instrumentation and methodology. The ongoing development of faster, more sensitive, and more specific GC-MS methods addresses critical forensic needs while maintaining the rigorous standards required for legal admissibility. As forensic challenges grow increasingly complex with the emergence of novel psychoactive substances and sophisticated chemical evidence, GC-MS methodologies will continue to provide the scientific foundation for reliable forensic chemical analysis and judicial decision-making.
From Crime Scene to Courtroom: Method Development and Forensic Applications of GC-MS
Rapid Screening Protocols for Seized Drugs and Toxicological Substances
Gas Chromatography-Mass Spectrometry (GC-MS) has long been the cornerstone of confirmatory analysis in forensic drug chemistry. However, conventional GC-MS techniques often require extensive analysis times, typically around 30 minutes per sample, creating significant bottlenecks in forensic laboratories dealing with escalating caseloads of seized drugs and toxicological substances [44]. The imperative for judicial and law enforcement agencies to receive timely analytical results has driven the development and validation of rapid GC-MS protocols that maintain the analytical rigor of traditional methods while dramatically increasing throughput. This technical guide examines the fundamental principles, methodologies, and applications of these advanced screening protocols within the broader context of GC-MS forensic research.
The evolution of rapid GC-MS technologies represents a paradigm shift in forensic analytical chemistry, addressing critical needs for expedited judicial processes and more responsive law enforcement operations. By optimizing temperature programming, carrier gas flow rates, and column parameters, modern rapid GC-MS methods can reduce total analysis time from approximately 30 minutes to as little as 10 minutes while maintaining—and in some cases enhancing—the sensitivity and specificity required for forensic applications [44]. These advancements are particularly valuable for laboratories facing substantial case backlogs and those processing large volumes of seized materials in time-sensitive investigations.
Key Advancements in Rapid GC-MS Methodology
Core Technological Innovations
The transition from conventional to rapid GC-MS screening has been enabled by several critical technological improvements. Method optimization has focused primarily on enhanced temperature programming and operational parameter adjustments that efficiently shorten run times without compromising chromatographic resolution or detection capabilities [44]. Specifically, researchers have implemented steeper temperature ramps (increased from 15°C/min to 70°C/min) and higher initial oven temperatures (raised from 70°C to 120°C), significantly accelerating the elution of analytes while maintaining effective separation [44].
Instrumental modifications have further contributed to these efficiency gains. The implementation of shorter and narrower chromatographic columns has demonstrated particular utility for reducing analysis time for seized drug samples [44]. When combined with increased carrier gas flow rates (optimized at 2 mL/min compared to 1 mL/min in conventional methods), these approaches facilitate faster analyte transport through the system while preserving the mass spectral quality essential for confident compound identification [44]. The resulting methods maintain the same 30-m DB-5 ms column format used in traditional analyses, ensuring compatibility with existing laboratory infrastructure and methodologies [44].
Experimental Protocol: Rapid GC-MS Screening
The following section details a standardized methodology for the rapid GC-MS screening of seized drugs and toxicological substances, compiled from validated forensic protocols [44] [45].
Instrument Configuration and Parameters
- Gas Chromatograph: Agilent 7890B system or equivalent
- Mass Spectrometer: Agilent 5977A single quadrupole mass spectrometer or equivalent
- Chromatographic Column: Agilent J&W DB-5 ms (30 m × 0.25 mm × 0.25 μm)
- Carrier Gas: Helium (99.999% purity) at a fixed flow rate of 2.0 mL/min
- Injection System: Split injection (20:1 ratio) at 280°C
- Temperature Program:
- Initial temperature: 120°C
- Ramp rate: 70°C/min to 300°C
- Final hold time: 7.43 minutes
- Total run time: 10.00 minutes
- MSD Parameters:
- Ionization mode: Electron Ionization (70 eV)
- Ion source temperature: 230°C
- Transfer line temperature: 280°C
- Quadrupole temperature: 150°C
- Mass scan range: m/z 40-550
Sample Preparation Procedure
- Stock Solution Preparation: Prepare individual reference standard solutions at approximately 1.0 mg/mL in HPLC-grade methanol.
- Working Standard Mixtures: Combine appropriate aliquots of stock solutions to create working mixtures containing target analytes at approximately 0.05 mg/mL per compound [44].
- Sample Extraction:
- For suspected controlled substances in powder form: Accurately weigh approximately 1-2 mg of material into a glass vial.
- Add 1.0 mL of methanol and vortex mix for 30 seconds.
- Centrifuge at 10,000 rpm for 5 minutes to sediment particulate matter.
- Transfer supernatant to GC vial for analysis.
- Quality Control: Include system suitability checks and continuing calibration verification samples in each analytical batch.
Workflow Visualization
The following Graphviz diagram illustrates the complete analytical workflow for rapid GC-MS screening of seized drugs:
Figure 1: Rapid GC-MS Analytical Workflow for Seized Drug Analysis
Performance Metrics and Validation Data
Quantitative Performance Assessment
Comprehensive validation studies have demonstrated that optimized rapid GC-MS methods significantly outperform conventional approaches across multiple performance metrics [44] [45]. The following tables summarize key quantitative data from validation studies conducted using real-case samples from forensic laboratories.
Table 1: Comparative Analytical Performance of Conventional vs. Rapid GC-MS Methods [44]
| Performance Parameter | Conventional GC-MS | Rapid GC-MS | Improvement |
|---|---|---|---|
| Total Analysis Time | 30.33 minutes | 10.00 minutes | 67% reduction |
| Cocaine LOD | 2.5 μg/mL | 1.0 μg/mL | 60% improvement |
| Heroin LOD | >2.0 μg/mL | ≤1.0 μg/mL | ≥50% improvement |
| Retention Time RSD | <1.0% | <0.25% | >75% improvement |
| Match Quality Scores | 85-90% | >90% | Consistent improvement |
Table 2: Method Validation Summary Across Multiple Drug Classes [44] [45]
| Validation Component | Acceptance Criteria | Experimental Results | Compliance Status |
|---|---|---|---|
| Selectivity | Differentiation of target analytes | Successful for most compounds; limitations with some isomers | Partial |
| Precision (RT RSD) | ≤10% | ≤0.25% for stable compounds | Meets criteria |
| Precision (Spectral RSD) | ≤10% | ≤10% for most compounds | Meets criteria |
| Accuracy | Correct identification in case samples | >90% match quality across concentrations | Meets criteria |
| Carryover/Contamination | <1% of LOD | Not detected in blank injections | Meets criteria |
| Robustness | Consistent performance under variation | % RSDs ≤10% under modified conditions | Meets criteria |
| Stability | Consistent response over 72h | <5% deviation in system response | Meets criteria |
Validation Framework and Acceptance Criteria
The implementation of rapid GC-MS protocols in forensic laboratories requires rigorous validation to establish method reliability and define performance boundaries. Recent research has developed comprehensive validation templates specifically designed for rapid GC-MS seized drug screening applications [45] [46]. These frameworks assess nine critical validation components: selectivity, matrix effects, precision, accuracy, range, carryover/contamination, robustness, ruggedness, and stability [45].
Validation studies have demonstrated that rapid GC-MS methods consistently achieve relative standard deviations (% RSDs) for retention times and mass spectral search scores of ≤10% for both precision and robustness studies, meeting the stringent acceptance criteria employed by accredited forensic laboratories [45]. The technique has shown particular effectiveness in real-case scenarios, with applications to authentic samples from forensic laboratories accurately identifying diverse drug classes—including synthetic opioids, stimulants, and cannabinoids—with match quality scores consistently exceeding 90% across tested concentrations [44].
The following Graphviz diagram illustrates the comprehensive validation framework for rapid GC-MS methods:
Figure 2: Rapid GC-MS Method Validation Framework
The Scientist's Toolkit: Essential Research Reagents and Materials
The successful implementation of rapid GC-MS screening protocols requires specific reagents, reference materials, and analytical resources. The following table details essential components of the forensic chemist's toolkit for seized drug analysis.
Table 3: Essential Research Reagents and Materials for Rapid GC-MS Drug Screening [44] [45]
| Item | Specification | Application/Purpose |
|---|---|---|
| Chromatographic Column | Agilent J&W DB-5 ms (30 m × 0.25 mm × 0.25 μm) | Primary separation column for drug compounds |
| Certified Reference Standards | Sigma-Aldrich (Cerilliant), Cayman Chemical | Target analyte identification and quantification |
| Solvents | HPLC-grade methanol, acetonitrile | Sample preparation, dilution, and extraction |
| Internal Standards | Deuterated analogs of target drugs | Quantitation and quality control |
| General Analysis Mixtures | Custom mixtures of common drugs (0.05-0.25 mg/mL per compound) | System suitability testing and method development |
| Mass Spectral Libraries | Wiley Spectral Library, Cayman Spectral Library | Compound identification and verification |
| Quality Control Materials | Positive and negative controls | Method validation and ongoing quality assurance |
| Data Processing Software | Agilent MassHunter, Enhanced ChemStation | Data acquisition, processing, and reporting |
Applications in Forensic Contexts
The practical implementation of rapid GC-MS screening has demonstrated significant utility across diverse forensic scenarios. In operational forensic laboratories, these methods have been successfully applied to the analysis of authentic case samples, including complex mixtures of synthetic opioids, stimulants, benzodiazepines, and novel psychoactive substances [44]. The technique's robust performance with real-world samples—including those with complex matrices—underscores its practical value in evidentiary analysis.
The efficiency gains achieved through rapid GC-MS protocols directly address the critical need for reduced forensic backlogs, facilitating faster law enforcement responses and judicial processes without compromising analytical confidence [44]. This balance of speed and reliability makes these methods particularly valuable for high-throughput laboratory environments and time-sensitive investigations where rapid intelligence gathering is essential for public safety interventions.
Rapid GC-MS screening protocols represent a significant advancement in forensic analytical chemistry, offering a validated approach to substantially decrease analysis time while maintaining the rigorous performance standards required for evidentiary applications. Through optimized instrument parameters, comprehensive validation frameworks, and standardized operating procedures, these methods deliver reproducible, reliable results for the identification of seized drugs and toxicological substances. The ongoing development and refinement of these protocols continue to enhance the capabilities of forensic laboratories worldwide, supporting more efficient administration of justice and effective public safety interventions.
Within the framework of gas chromatography-mass spectrometry (GC-MS) forensics research, the mastery of sample preparation is a foundational prerequisite for generating legally defensible and analytically sound results. This technical guide provides an in-depth examination of extraction and derivatization techniques specifically tailored for complex matrices encountered in forensic laboratories. We detail systematic methodologies for isolating target analytes from challenging sample types and modifying their chemical structures to enhance detectability, with a focused application to forensic science. The protocols and data presented herein are designed to equip researchers and drug development professionals with advanced practical knowledge for improving accuracy, sensitivity, and reliability in GC-MS analyses, thereby strengthening the evidentiary value of analytical findings in forensic investigations.
Gas chromatography-mass spectrometry (GC-MS) combines two powerful analytical techniques to separate, identify, and quantify chemical substances found in complex sample matrices [47]. The gas chromatograph separates volatile components in a mixture through a temperature-controlled column, while the mass spectrometer detects and identifies the separated compounds based on their mass-to-charge ratio (m/z) [5]. This hyphenated technique provides three-dimensional data—retention time, response intensity, and mass spectral information—that serves as a forensic fingerprint for compound identification [5].
In forensic contexts, proper sample preparation is paramount as evidentiary materials often present in complex, dirty, or biologically variable matrices such as blood, urine, tissue, seized drug compounds, and environmental samples [48]. Without adequate preparation, these matrices can interfere with analysis, damage instrumentation, or generate misleading results that compromise forensic conclusions [48]. Effective sample preparation ensures that target analytes are isolated, concentrated, and presented to the GC-MS system in an optimal state, thereby enhancing method accuracy, reproducibility, and sensitivity while protecting instrumental components [49] [48].
Fundamental Extraction Techniques
Extraction represents the critical first step in sample preparation, aiming to isolate target analytes from complex matrices while minimizing interfering substances. The selection of an appropriate extraction technique depends on the sample composition, target analyte characteristics, and required sensitivity.
Solid Phase Extraction (SPE)
Solid Phase Extraction utilizes cartridges containing a solid packing material that, in combination with selective solvent systems, allows for the separation of target analytes from the sample matrix based on chemical interactions [49]. The selection of SPE sorbent chemistry must align with the characteristics of the target analytes, as illustrated in Figure 2 of the search results [49]. For instance, reversed-phase SPE (C8, C18) suits non-polar compounds, while normal-phase (silica, alumina) or ion-exchange phases target polar or ionic analytes respectively [49].
Experimental Protocol: Solid Phase Extraction for Biological Fluids
- Conditioning: Activate the SPE cartridge (e.g., C18 for reversed-phase extraction) with 5-10 mL of methanol followed by 5-10 mL of deionized water or buffer at a flow rate of 1-2 mL/min.
- Loading: Apply the prepared biological sample (e.g., urine, blood supernatant) to the cartridge at a controlled flow rate of 1-3 mL/min, ensuring target analytes are retained while interfering matrix components pass through.
- Washing: Remove weakly retained interferences with 5-10 mL of a weak solvent (e.g., 5% methanol in water for reversed-phase SPE).
- Elution: Recover the target analytes with 5-10 mL of a strong solvent (e.g., pure methanol or acetonitrile for reversed-phase SPE), collecting the eluate in a clean vessel.
- Concentration: Evaporate the eluate to dryness under a gentle stream of nitrogen or using a vacuum concentrator, then reconstitute in an appropriate solvent compatible with GC-MS analysis [49] [48].
QuEChERS
The QuEChERS (Quick, Easy, Cheap, Effective, Rugged, and Safe) methodology represents a streamlined approach to sample preparation that has gained significant traction in forensic and food safety laboratories for its efficiency in processing complex matrices [49]. This technique employs a salting-out extraction followed by a dispersive solid-phase extraction (d-SPE) clean-up step to efficiently isolate analytes while reducing matrix effects.
Experimental Protocol: QuEChERS for Complex Matrices
- Extraction: Weigh 10-15 g of homogenized sample into a 50 mL centrifuge tube. Add 10-15 mL of acetonitrile and shake vigorously for 1 minute. Incorporate a salt mixture (typically containing magnesium sulfate and sodium chloride) to induce phase separation and shake again.
- Centrifugation: Centrifuge the mixture at 3000-5000 rpm for 5 minutes to achieve clear phase separation.
- Clean-up: Transfer an aliquot (e.g., 1 mL) of the upper organic layer to a d-SPE tube containing clean-up sorbents (typically primary-secondary amine (PSA) for pigment removal and C18 for lipid elimination).
- Shaking and Centrifugation: Vortex the d-SPE tube for 30-60 seconds, then centrifuge at 3000-5000 rpm for 5 minutes.
- Preparation for Analysis: Transfer the purified extract to an autosampler vial for direct analysis or proceed with derivatization if required [49].
Liquid-Liquid Extraction (LLE)
Liquid-Liquid Extraction separates compounds based on their differential solubility in two immiscible liquids, typically an organic solvent and an aqueous phase [48]. This technique remains widely employed in forensic toxicology for its effectiveness in extracting a broad spectrum of analytes from biological matrices.
Experimental Protocol: Liquid-Liquid Extraction for Basic Drugs
- Sample Preparation: Adjust the pH of the biological sample (e.g., blood, urine) to approximately 9 using ammonium hydroxide or buffer to ensure basic drugs remain in their non-ionized form.
- Extraction: Add 5-10 mL of appropriate organic solvent (e.g., ethyl acetate, chloroform, or hexane) to the sample in a separation funnel or centrifuge tube. Shake vigorously for 2-5 minutes, periodically venting pressure.
- Phase Separation: Allow the phases to separate naturally or accelerate separation by centrifugation at 3000 rpm for 5 minutes.
- Collection: Transfer the organic (upper) layer containing the extracted analytes to a clean evaporation vessel.
- Concentration: Evaporate the organic extract to dryness under a gentle nitrogen stream at 30-40°C. Reconstitute the residue in 50-200 µL of appropriate solvent for GC-MS analysis [48].
Table 1: Comparison of Major Extraction Techniques for Forensic Applications
| Technique | Principles | Optimal Applications | Advantages | Limitations |
|---|---|---|---|---|
| Solid Phase Extraction (SPE) | Partitioning between solid sorbent and liquid mobile phase | Biological fluids, environmental water samples, purified drug extracts | High selectivity, efficient clean-up, automation compatibility | Method development complexity, cartridge cost |
| QuEChERS | Salting-out extraction with d-SPE clean-up | Food commodities, plant materials, biological tissues | Rapid processing, minimal solvent usage, high throughput | Limited effectiveness for very polar compounds |
| Liquid-Liquid Extraction (LLE) | Differential solubility in immiscible liquids | Broad-spectrum drug screening, liquid samples | Simplicity, well-established methods, no specialized equipment | Emulsion formation, large solvent volumes, manual intensive |
Advanced Derivatization Strategies
Derivatization chemically modifies target analytes to enhance their suitability for GC-MS analysis by improving volatility, thermal stability, and detectability [49]. This process is particularly crucial in forensic analysis where target compounds may contain functional groups that render them poorly compatible with GC-MS in their native form.
Solid-Phase Analytical Derivatization (SPAD)
Solid-Phase Analytical Derivatization represents an innovative approach that combines the principles of solid-phase extraction with analytical derivatization in a single integrated workflow [50]. This technique provides significant advantages in analysis accuracy, efficiency, reproducibility, and sensitivity by concentrating analytes and derivatizing reagents in a confined environment [50].
Experimental Protocol: SPAD for Carbonyl Compounds
- SPE Column Preparation: Pre-condition an appropriate SPE cartridge (e.g., C18 for reversed-phase) with methanol and water.
- Reagent Loading: Impregnate the solid phase with a derivatizing reagent solution (e.g., pentafluorobenzylhydroxylamine for aldehydes and ketones) in a volatile organic solvent, then remove excess solvent under gentle vacuum.
- Sample Application: Pass the prepared sample extract through the derivatization-activated SPE cartridge, allowing simultaneous extraction and derivatization to occur.
- Washing: Remove unreacted reagents and matrix interferences by passing a suitable wash solvent through the cartridge.
- Elution: Recover the derivatized analytes using a strong elution solvent (e.g., dichloromethane or ethyl acetate).
- Analysis: Directly analyze the eluate by GC-MS without additional treatment [50].
Common Derivatization Approaches
The selection of derivatization reagents and methods depends primarily on the functional groups present in the target analytes and the specific analytical requirements. The following table summarizes principal derivatization approaches relevant to forensic analysis.
Table 2: Derivatization Reagents and Applications in Forensic GC-MS
| Derivatization Type | Common Reagents | Target Functional Groups | Forensic Applications | Key Advantages |
|---|---|---|---|---|
| Silylation | BSTFA, MSTFA, TMSI | -OH, -COOH, -NH, -SH | Cannabis analysis, steroid profiling, drug metabolites | Broad applicability, high volatility derivatives, commercial availability |
| Acylation | TFAA, HFBA, MBTFA | -OH, -NH₂ | Amphetamines, catecholamines, aminoglycosides | Enhanced electron capture detection, improved chromatographic performance |
| Alkylation | MTBSTFA, PFBBr, DMA | -COOH, -OH | Carboxylic acids, barbiturates, fatty acids | Increased thermal stability, mass spectral characteristics |
| Condensation | Methoxyamine, pentafluorobenzylhydroxylamine | C=O (carbonyl) | Aldehydes, ketones, keto-acids | Specificity for carbonyl compounds, enhanced mass spectral properties |
Experimental Protocol: Silylation for Hydroxyl-Containing Compounds
- Sample Preparation: Ensure the extracted sample is completely dry, as water interferes with silylation reactions.
- Reagent Addition: Add 50-100 µL of silylation reagent (e.g., N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) with 1% trimethylchlorosilane (TMCS)) to the dried extract.
- Reaction Incubation: Heat the mixture at 60-80°C for 20-45 minutes to complete the derivatization reaction.
- Direct Analysis: Transfer the reaction mixture directly to a GC vial for analysis. Excess reagent typically does not interfere with chromatography and can be vented in the GC inlet [49] [50].
Integrated Workflows for Forensic Specimens
The effective integration of extraction and derivatization techniques into cohesive workflows represents the pinnacle of sample preparation mastery in forensic GC-MS analysis. The following diagram illustrates a generalized workflow for processing complex forensic specimens, highlighting critical decision points and technique selection criteria.
Diagram 1: Integrated workflow for forensic sample preparation in GC-MS analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of extraction and derivatization protocols requires access to specialized reagents and materials. The following table catalogs essential components of the sample preparation toolkit for forensic GC-MS applications.
Table 3: Essential Research Reagent Solutions for Forensic Sample Preparation
| Reagent/Material | Function | Application Examples | Technical Notes |
|---|---|---|---|
| C18 SPE Cartridges | Reversed-phase extraction medium | Extraction of non-polar to moderately polar analytes from biological fluids | Various bed weights (100-500 mg) and cartridge sizes available for different sample loads |
| Mixed-Mode SPE Cartridges | Combined reversed-phase and ion-exchange mechanisms | Simultaneous extraction of acidic, basic, and neutral compounds | Particularly valuable for comprehensive drug screening in forensic toxicology |
| Primary-Secondary Amine (PSA) | Removal of polar interferences (acids, pigments, sugars) | QuEChERS clean-up of food, plant, and biological matrices | Effective for removing fatty acids and other co-extracted polar matrix components |
| BSTFA with 1% TMCS | Silylation derivatizing reagent | Derivatization of hydroxyl, carboxyl, and amine groups in drugs and metabolites | TMCS acts as a catalyst; reaction must be performed under anhydrous conditions |
| Pentafluorobenzylhydroxylamine (PFBHA) | Oxime formation with carbonyl compounds | Derivatization of aldehydes and ketones for enhanced EI-MS detection | Imparts strong electron-capturing properties improving sensitivity in SIM modes |
| MSTFA | Silylation reagent for active hydrogens | Derivatization of steroids, cannabinoids, and metabolic profiling | Produces volatile derivatives with excellent chromatographic properties |
| Anhydrous Pyridine | Reaction solvent and acid acceptor | Derivatization reactions, particularly acylation and silylation | Must be stored with molecular sieves to maintain anhydrous conditions |
| N-Methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA) | Tert-butyldimethylsilylation reagent | Derivatization of compounds requiring exceptionally stable derivatives | TBDMS derivatives exhibit characteristic fragmentation patterns with m/z 57 |
Mastery of extraction and derivatization techniques represents a cornerstone of effective GC-MS analysis in forensic research. Through strategic selection and implementation of appropriate sample preparation methodologies—ranging from conventional SPE and LLE to innovative approaches like QuEChERS and SPAD—analysts can significantly enhance the quality and reliability of forensic data. The integrated workflows and detailed protocols presented in this guide provide a structured framework for addressing the analytical challenges posed by complex matrices typically encountered in forensic investigations. As the field continues to evolve, ongoing refinement of these sample preparation strategies will remain essential for advancing the scientific rigor of forensic chemistry and strengthening the evidentiary foundation of analytical findings in legal contexts.
In the realm of forensic trace evidence, microscopic fibers represent one of the most prevalent and valuable forms of transferable material encountered at crime scenes. Due to their high transfer potential, fibers can provide crucial associative evidence linking individuals to specific locations, victims, or objects [12]. However, forensic fiber analysis faces substantial challenges—evidential fibers recovered from crime scenes are typically extremely small, often measuring only a few millimeters in length with diameters below 25 μm, while the dye content within a single fiber is proportionally minute, estimated at a mere 2-200 nanograms [12]. Among synthetic fibers, polyester dominates the global market, accounting for approximately 57% of total fiber production worldwide, with azo dyes constituting up to 70% of the colorants used in their coloration [12]. This prevalence makes the analysis of azo dyes in polyester fibers particularly significant for forensic discrimination.
Traditional analytical approaches for fiber examination have primarily relied on microscopic and spectroscopic techniques, including microspectrophotometry (MSP) and Raman spectroscopy [51]. While these methods provide valuable preliminary data, they often lack the specificity required to discriminate between fibers dyed with different chemical compositions that may appear visually similar. This limitation has driven the development of more sophisticated chromatographic methods that can identify specific dye components, thereby enhancing the evidential value of fiber comparisons [52]. The integration of gas chromatography with tandem mass spectrometry (GC-MS/MS) represents a particularly innovative approach, leveraging the structural characteristics of azo dyes to enable highly sensitive and selective analysis of trace fiber evidence.
Fundamental Principles: Azo Dyes and GC-MS/MS Analysis
Structural Characteristics of Azo Dyes
Azo dyes constitute the largest and most structurally diverse class of synthetic dyes, accounting for approximately 60-65% of all commercial dye production worldwide [12] [53]. These compounds are characterized by the presence of at least one azo group (–N=N–) connecting aromatic systems, which serves as the chromophore responsible for their color properties. The extensive conjugation between the azo bond and adjacent aromatic structures allows these molecules to absorb light in the visible spectrum, producing vibrant colors that make them ideal for textile applications. From a forensic perspective, this structural complexity becomes highly valuable for discrimination purposes, as manufacturers utilize numerous dye combinations and ratios to achieve specific color shades, creating chemically distinct signatures that can differentiate otherwise similar-appearing fibers.
The forensic analysis of azo dyes presents unique challenges due to their molecular characteristics. Many disperse dyes used for polyester fibers have high boiling points and limited volatility, creating potential difficulties for GC-based analysis [12]. However, this challenge can be circumvented through chemical modification—specifically, the reductive cleavage of the azo bond to produce smaller, more volatile aromatic amines that are amenable to gas chromatographic separation [12]. This strategic approach transforms the analytical problem from dealing with complex, high-molecular-weight dyes to identifying their constituent amine fragments, which serve as molecular markers for the original dye compounds present in the fiber.
GC-MS/MS Fundamentals in Forensic Context
Gas chromatography coupled with tandem mass spectrometry (GC-MS/MS) represents a powerful analytical platform for forensic fiber analysis, combining high separation efficiency with exceptional detection specificity and sensitivity. The GC component separates complex mixtures of volatile compounds based on their differential partitioning between a mobile gas phase and a stationary liquid phase within a chromatographic column. This separation is particularly crucial for fiber extracts, which may contain multiple dye components and potential interferents from the fiber matrix or environmental contamination.
The tandem mass spectrometry component provides two stages of mass analysis separated by a collision cell, enabling selective detection of target analytes even in complex matrices. When operated in Multiple Reaction Monitoring (MRM) mode, the instrument specifically monitors predetermined precursor-product ion transitions for each target compound, significantly enhancing signal-to-noise ratios and method sensitivity compared to full-scan acquisition techniques [12]. This capability is paramount for forensic fiber analysis, where analyte concentrations are extremely limited. The MRM approach provides two independent identification parameters—retention time and specific ion transitions—that together create a highly specific analytical signature for each target amine, reducing the likelihood of false positives and increasing the reliability of forensic conclusions [12].
Experimental Workflow for Azo Dye Analysis in Trace Fibers
Comprehensive Analytical Protocol
The complete analytical procedure for discriminating polyester fibers based on their azo dye content encompasses five critical stages: dye extraction from fibers, reductive cleavage of azo bonds, isolation of resulting aromatic amines, identification via GC-MS/MS analysis, and final fiber discrimination based on detected amine profiles [12]. This systematic approach transforms the complex dye molecules into more analytically tractable compounds while maintaining the chemical signature necessary for discrimination.
Detailed Methodological Procedures
Dye Extraction from Polyester Fibers
The extraction process represents a critical first step in recovering dye molecules from the polyester matrix. Polyester fibers and disperse dyes are both hydrophobic, necessitating the use of appropriate organic solvents for efficient extraction [51]. Based on optimization studies, chlorobenzene has been identified as the most effective extraction solvent, achieving nearly complete recovery of dye compounds from polyester fibers [12] [51]. The standard extraction protocol involves placing a single 2 cm fiber segment into a glass capillary tube with 5 μL of chlorobenzene, sealing the tube, and heating at 100°C for 15 minutes [51]. This minimal-volume approach is specifically designed to maintain adequate analyte concentration for subsequent analysis while working within the physical constraints of single-fiber evidence.
Reductive Cleavage of Azo Bonds
Following extraction, the azo dyes undergo chemical reduction to break the azo bonds and generate their constituent aromatic amines. This process adapts and miniaturizes the standard procedure described in EN ISO 14362-1:2017, which originally required gram quantities of textile material [12]. The modified forensic protocol utilizes sodium dithionite (Na₂S₂O₄) as the reducing agent in a citrate buffer system (pH ≈ 6) at 70°C for 10 minutes [12]. This critical transformation enables subsequent GC-MS/MS analysis by converting the relatively non-volatile dye compounds into smaller, more volatile aromatic amines that are amenable to gas chromatographic separation. The reduction reaction follows a straightforward mechanism where the azo bond cleaves to generate two amine products, as illustrated below:
Aromatic Amine Isolation via DLLME
Dispersive Liquid-Liquid Microextraction (DLLME) follows reductive cleavage to concentrate the aromatic amines and remove potential interferents prior to instrumental analysis [12]. In this efficient microextraction technique, a mixture of extraction solvent (chloroform) and disperser solvent (acetonitrile) is rapidly injected into the aqueous sample containing the aromatic amines. This creates a cloudy solution consisting of fine droplets of extraction solvent dispersed throughout the aqueous phase, providing an enormous surface area for rapid partitioning of the target analytes from the aqueous phase into the organic phase. The solution is then centrifuged to sediment the dense extraction solvent droplets at the bottom of the tube, and a predetermined volume of the sedimented phase is collected for GC-MS/MS analysis. DLLME provides exceptional preconcentration factors and cleanup efficiency, making it ideally suited for the minute sample quantities available in forensic fiber analysis.
GC-MS/MS Instrumental Parameters
The final analytical separation and detection employ optimized GC-MS/MS conditions specifically developed for the target aromatic amines derived from azo dye reduction. The analysis utilizes a gas chromatograph equipped with a (5%-phenyl)-methylpolysiloxane capillary column (30 m × 250 μm × 0.25 μm) coupled to a tandem mass spectrometer [12] [54]. The GC oven temperature program initiates at 70°C, then ramps to 130°C at 17°C/min (held for 1 minute), followed by a second ramp to 290°C at 50°C/min (held for 2.27 minutes) [54]. Helium serves as the carrier gas at a constant flow rate of 1.8 mL/min [54].
The mass spectrometer operates in Multiple Reaction Monitoring (MRM) mode with electron ionization (70 eV) [12] [54]. This highly specific detection mode monitors predetermined precursor-product ion transitions for each target amine, significantly enhancing method sensitivity and selectivity compared to full-scan acquisition techniques. The MRM approach provides two independent identification parameters—retention time and specific ion transitions—that together create a highly specific analytical signature for each target amine [12].
Analytical Performance and Validation Data
Quantitative Method Performance
The developed GC-MS/MS method demonstrates excellent analytical performance characteristics suitable for forensic applications. Method validation studies have established good interday precision, with coefficients of variation (CV%) ranging from 0.03–0.18% for retention times and 3.27–21.03% for peak areas across the target aromatic amines [12]. These precision metrics indicate sufficient method robustness for reliable compound identification and semi-quantitative comparison of fiber samples.
Table 1: Analytical Performance Metrics of GC-MS/MS Method for Azo Dye Analysis
| Performance Parameter | Performance Range | Significance in Forensic Analysis |
|---|---|---|
| Retention Time Precision | 0.03–0.18% CV | Ensures reliable compound identification through consistent retention times |
| Peak Area Precision | 3.27–21.03% CV | Enables semi-quantitative comparison of dye components between samples |
| Discriminatory Power | 87% | Demonstrates high effectiveness in distinguishing between different fiber sources |
| Fiber Sample Length | 2 cm | Compatible with typical evidence sample sizes encountered in casework |
Forensic Discrimination Efficacy
The practical utility of the methodology was evaluated using 22 real-world samples consisting of 2 cm-long single fibers collected from various garments [12]. The analysis achieved a discriminatory power of 87%, confirming the method's robustness and potential utility in forensic applications [12]. This high discrimination rate signifies that the approach can effectively distinguish between fibers originating from different sources based on their azo dye profiles, thereby providing meaningful associative evidence for criminal investigations.
The method offers two distinct operational approaches depending on the available reference materials. When abundant reference material is available (e.g., 5 cm thread), a selective method can be developed where dyes are first identified in SCAN mode, followed by MRM optimization for specific target ions [12]. For cases where both evidence and reference samples are limited to single fibers, a universal screening method can be employed based on predetermined MRM transitions for the most frequently encountered aromatic amines in polyester fibers [12]. This operational flexibility makes the technique adaptable to various evidentiary scenarios commonly encountered in forensic practice.
Research Reagent Solutions and Essential Materials
Successful implementation of the azo dye analysis workflow requires specific reagents and materials optimized for each procedural step. The following table comprehensively details the essential research reagents and their specific functions within the analytical protocol:
Table 2: Essential Research Reagents and Materials for Azo Dye Analysis in Trace Fibers
| Reagent/Material | Specific Function | Application Notes |
|---|---|---|
| Chlorobenzene | Extraction solvent for disperse dyes from polyester fibers | High efficiency for hydrophobic dyes; used at 100°C for 15 minutes [12] [51] |
| Sodium Dithionite (Na₂S₂O₄) | Reducing agent for azo bond cleavage | Generates aromatic amines from azo dyes at 70°C in citrate buffer [12] |
| Citrate Buffer (pH ≈ 6) | Reaction medium for reductive cleavage | Maintains optimal pH for the reduction reaction [12] |
| Chloroform | Extraction solvent in DLLME | High density for easy sediment separation; efficiently extracts aromatic amines [12] |
| Acetonitrile | Disperser solvent in DLLME | Enhances formation of fine chloroform droplets in aqueous solution [12] |
| (5%-phenyl)-methylpolysiloxane GC Column | Stationary phase for chromatographic separation | Provides optimal separation of aromatic amine derivatives [12] [54] |
Comparative Analytical Techniques and Advancements
Alternative Methodologies in Forensic Dye Analysis
While the GC-MS/MS approach for azo dye analysis offers significant advantages, several alternative methodologies have been developed for forensic fiber examination. High-performance liquid chromatography (HPLC) with mass spectrometric detection has been extensively utilized for the direct analysis of disperse dyes without requiring chemical reduction [51]. HPLC methods typically achieve separation times ranging from 20 to 67 minutes, though the implementation of ultra high-performance liquid chromatography (UHPLC) can reduce this to approximately 10 minutes [12]. Liquid chromatography coupled with diode array detection and mass spectrometry (HPLC-DAD-MS) has been successfully applied to various fiber types including cotton, polyester, and polyamide, enabling the identification of dye mixtures in fiber traces [52].
More recently, innovative approaches such as automated microfluidics extraction systems coupled with Q-TOF mass spectrometers have emerged, enabling dye characterization from single 1 mm fiber segments with total analysis times under 12 minutes [55]. Direct mass spectrometric techniques, including infrared matrix-assisted laser desorption electrospray ionization, have also shown promise for minimal sample preparation approaches [51]. Each methodology presents distinct advantages and limitations regarding sensitivity, specificity, analysis time, instrumental requirements, and applicability to different fiber-dye systems.
Advantages of GC-MS/MS for Azo Dye Analysis
The GC-MS/MS approach for analyzing azo dyes through their aromatic amine derivatives offers several distinctive benefits for forensic fiber discrimination. A significant practical advantage is the widespread availability of GC-MS instrumentation in forensic laboratories compared to more specialized techniques such as UHPLC or UHPSFC [12]. This accessibility enhances the method's potential for adoption into routine casework. The MRM detection mode provides exceptional sensitivity and selectivity, enabling successful analysis of single 2 cm fiber segments comparable to evidence samples encountered at crime scenes [12].
The chemical reduction step transforms the analytical challenge from dealing with diverse, high-molecular-weight dye structures to targeting a defined set of aromatic amines, creating a more standardized approach that can be applied across different dye formulations. The demonstrated discriminatory power of 87% confirms the method's effectiveness for distinguishing between fibers of similar color but different dye compositions [12]. Furthermore, the technique complements existing microscopic and spectroscopic methods, adding a chemical dimension to fiber comparisons that enhances the overall evidential value.
The analysis of azo dyes in trace fibers through GC-MS/MS represents a significant advancement in forensic fiber discrimination methodology. By leveraging the structural characteristics of azo dyes and transforming them into analytically tractable aromatic amine derivatives, this approach overcomes the challenges associated with the direct analysis of complex dye molecules in minute fiber samples. The comprehensive workflow encompassing dye extraction, reductive cleavage, DLLME concentration, and GC-MS/MS analysis provides a robust framework for obtaining chemically specific discrimination between visually similar fibers.
The method's validation with real-world samples and demonstrated discriminatory power of 87% confirm its practical utility for forensic casework [12]. Furthermore, the technique's compatibility with standard GC-MS instrumentation present in most forensic laboratories enhances its potential for widespread adoption. As synthetic fibers and complex dye formulations continue to evolve in the textile industry, such chemically specific analytical approaches will become increasingly vital for maximizing the evidential value of fiber traces in criminal investigations. The methodology not only complements existing techniques but also provides a foundational approach that can be adapted and expanded to address new analytical challenges in forensic fiber examination.
The analysis of intoxicants in decomposed viscera represents one of the most complex challenges in forensic toxicology. During the decomposition process, biological tissues undergo significant chemical and structural changes, including autolysis, putrefaction, and the production of myriad volatile organic compounds that can interfere with analytical methods. These postmortem changes complicate the isolation, identification, and quantification of toxic substances, requiring sophisticated sample preparation and analytical techniques to ensure reliable results. Gas chromatography-mass spectrometry (GC-MS) has emerged as a cornerstone technology for addressing these challenges, providing the separation power and detection specificity needed for accurate toxicological analysis in forensic investigations [56] [57].
The complexity of decomposed tissue matrices necessitates robust analytical protocols that can effectively isolate target analytes from interfering substances while maintaining analytical integrity. Viscera samples from decomposed remains typically contain a complex mixture of proteins, lipids, degradation products, and microbial metabolites that can co-elute with compounds of interest, potentially causing false positives or negatives without adequate chromatographic separation. This technical guide outlines comprehensive methodologies for analyzing intoxicants in decomposed viscera using GC-MS, with detailed protocols, data presentation standards, and workflow visualizations designed specifically for forensic researchers and toxicology professionals working within the framework of basic GC-MS forensic research principles.
GC-MS Fundamentals for Forensic Analysis
Gas chromatography-mass spectrometry combines two powerful analytical techniques to provide high-resolution separation and definitive identification of chemical compounds. In GC-MS analysis, the gas chromatograph separates volatile and semi-volatile compounds in a complex mixture, while the mass spectrometer detects and identifies these compounds based on their mass-to-charge ratio (m/z) [56]. This combination is particularly valuable for forensic applications where evidentiary samples often contain numerous interfering substances that must be distinguished from target analytes.
The fundamental components of a GC-MS system include an injection port, chromatographic column, oven, interface, ion source, mass analyzer, and detector. Samples are introduced via the injection port, vaporized, and carried through the column by an inert gas mobile phase. Separation occurs based on differences in partitioning between the mobile phase and stationary phase within the column. As compounds elute from the column, they enter the mass spectrometer where they are ionized, typically by electron impact (EI) or chemical ionization (CI), and the resulting ions are separated by the mass analyzer before detection [56]. The mass spectrometer generates a fragmentation pattern that serves as a chemical fingerprint for each compound, enabling highly specific identification even in complex biological matrices like decomposed viscera.
For forensic applications, GC-MS offers several critical advantages, including the ability to analyze a wide range of compounds without extensive method development for each new sample, high confidence in analyte identification through mass spectral matching, and improved sensitivity for compounds that are otherwise challenging to detect [21] [56]. These characteristics make GC-MS particularly suitable for analyzing decomposed viscera, where the chemical background is complex and constantly changing throughout the decomposition process.
Sample Preparation Techniques for Complex Matrices
Tissue Homogenization and Extraction
Proper sample preparation is critical for successful analysis of intoxicants in decomposed viscera. The process begins with tissue homogenization, where approximately 1-2 grams of visceral tissue (liver, kidney, or brain tissue is often most informative for toxicological analysis) is weighed and homogenized in a suitable buffer solution, typically phosphate buffer (pH 7.4) or distilled water, at a ratio of 1:3 (tissue to buffer). The homogenization process should be conducted using a mechanical homogenizer for 2-3 minutes at high speed until a uniform consistency is achieved. For highly decomposed tissues, additional steps may be necessary, including filtration or centrifugation to remove particulate matter that could interfere with subsequent analysis.
Following homogenization, samples undergo extraction to isolate compounds of interest from the complex tissue matrix. Liquid-liquid extraction (LLE) remains a widely used technique, particularly for broad-spectrum toxicological analysis. A standardized LLE protocol involves:
- Transferring 2 mL of homogenate to a glass centrifuge tube
- Adding internal standard (appropriate deuterated analogs of target analytes)
- Adjusting pH to optimize compound recovery (alkaline pH ~9 for basic drugs, acidic pH ~2 for acidic compounds)
- Adding 5 mL of organic extraction solvent (ethyl acetate:hexane, 80:20 v/v)
- Vortex mixing for 3 minutes followed by centrifugation at 3500 rpm for 10 minutes
- Transferring the organic layer to a clean tube
- Repeating the extraction process and combining organic layers
- Evaporating to dryness under a gentle nitrogen stream at 40°C
- Reconstituting in 100 μL of appropriate solvent (methanol or mobile phase compatible solvent) for analysis
For more specific applications or challenging matrices, solid-phase extraction (SPE) provides enhanced clean-up and better recovery for certain compound classes. SPE protocols typically involve conditioning the sorbent (C18 or mixed-mode), sample loading, washing with aqueous buffer, and eluting with organic solvent. The choice between LLE and SPE depends on the target analytes, matrix complexity, and required sensitivity.
Derivatization Techniques
Many compounds of toxicological interest, particularly polar metabolites or compounds with active hydrogens, require chemical derivatization to improve their chromatographic behavior and detection sensitivity in GC-MS analysis. Derivatization serves to reduce polarity, increase volatility, and enhance thermal stability, which is particularly important for compounds that might otherwise degrade in the GC inlet or column. Common derivatization approaches include:
- Silylation: Using reagents like BSTFA (N,O-bis(trimethylsilyl)trifluoroacetamide) or MSTFA (N-methyl-N-trimethylsilyltrifluoroacetamide) to replace active hydrogens with trimethylsilyl groups, effective for alcohols, carboxylic acids, and amines.
- Acylation: Using reagents like TFAA (trifluoroacetic anhydride) or HFBA (heptafluorobutyric anhydride) to derivative amines and phenols, improving volatility and providing characteristic mass spectral fragments.
- Alkylation: Using reagents like TMAH (tetramethylammonium hydroxide) or iodomethane to derivative carboxylic acids.
The derivatization process typically involves reconstituting the dried extract in 50 μL of derivatization reagent, incubating at a specific temperature (often 60-70°C) for 15-30 minutes, and directly injecting into the GC-MS system. Proper derivatization is particularly important when analyzing decomposed viscera, as it can help distinguish target analytes from matrix interferences that may be present in the sample.
GC-MS Methodologies and Instrumental Parameters
GC-MS Configuration and Conditions
Optimal GC-MS analysis of intoxicants in decomposed viscera requires careful instrument configuration and parameter optimization to address the challenges posed by complex tissue matrices. The following table summarizes recommended instrumental conditions for routine analysis:
Table 1: Standard GC-MS Operating Conditions for Analysis of Intoxicants in Decomposed Viscera
| Parameter | Configuration/Setting | Alternative Options |
|---|---|---|
| GC System | Agilent 7890B | Equivalent performance systems |
| MS System | Agilent 5977B Quadrupole MSD | Other quadrupole mass spectrometers |
| Injection | Splitless mode (1 μL) | Pulsed splitless for better peak shape |
| Inlet Temperature | 250°C | 280°C for high boiling point compounds |
| Carrier Gas | Helium, constant flow 1.0 mL/min | Hydrogen (with safety precautions) |
| Oven Program | Initial 60°C (1 min), ramp 20°C/min to 320°C (10 min) | Modified for specific compound needs |
| Column | HP-5MS (30 m × 0.25 mm × 0.25 μm) | DB-5MS, Rxi-5Sil MS equivalent |
| Transfer Line | 280°C | 300°C for high molecular weight compounds |
| Ion Source | Electron Impact (70 eV), 230°C | Chemical ionization for specific applications |
| Quadrupole | 150°C | - |
| Solvent Delay | 3 minutes | Adjusted based on target analytes |
| Data Acquisition | Full scan (m/z 40-550) | SIM for improved sensitivity |
For particularly challenging samples where co-elution is problematic, comprehensive two-dimensional gas chromatography (GC×GC-MS) provides enhanced separation capability. GC×GC-MS employs two columns with different stationary phases connected through a modulator, dramatically increasing peak capacity and resolution [21]. This technique is particularly valuable for distinguishing target analytes from matrix interferences in decomposed viscera, where numerous biological compounds may co-elute with compounds of interest in conventional one-dimensional GC-MS.
Mass Spectrometric Detection Parameters
Mass spectrometric detection parameters must be optimized for the specific analytes of interest while maintaining the capability to detect unexpected compounds in forensic samples. Electron impact ionization (EI) at 70 eV is standard, providing reproducible mass spectra that can be matched against commercial libraries. The mass spectrometer should be tuned and calibrated according to manufacturer specifications using standard calibration compounds (e.g., perfluorotributylamine) to ensure mass accuracy and resolution.
Data acquisition can be performed in full scan mode for general unknown screening or selected ion monitoring (SIM) for targeted analysis with improved sensitivity. For full scan acquisition, a mass range of m/z 40-550 is typically sufficient to cover most toxicologically relevant compounds, with a scan rate of 2-5 scans per second to ensure adequate data points across chromatographic peaks. For SIM applications, characteristic ions for each target compound should be selected, typically 2-3 primary ions for quantification and confirmation, with dwell times optimized to maintain adequate points across the peak while maximizing sensitivity.
Quantitative Analysis and Method Validation
Calibration Approaches and Quality Control
Robust quantitative analysis requires establishment of a proper calibration model with appropriate quality control measures. A minimum of six calibration points should be used, spanning the expected concentration range in samples. For toxicological applications, this typically ranges from the limit of quantification to concentrations well above those expected in case samples. The following table presents validation parameters and acceptance criteria for quantitative GC-MS methods in forensic analysis:
Table 2: Method Validation Parameters and Acceptance Criteria for Quantitative GC-MS Analysis
| Validation Parameter | Recommended Procedure | Acceptance Criteria |
|---|---|---|
| Linearity | Minimum 6 calibration points | R² ≥ 0.99 |
| Accuracy | Quality controls at 3 levels | 85-115% of nominal value |
| Precision | Intra-day (n=6) and inter-day (n=18) | RSD ≤ 15% (≤20% at LLOQ) |
| Limit of Detection (LOD) | Signal-to-noise ratio ≥ 3:1 | Compound dependent |
| Lower Limit of Quantification (LLOQ) | Signal-to-noise ratio ≥ 10:1 | Accuracy 80-120%, RSD ≤ 20% |
| Recovery | Comparison with neat standards | Consistent and reproducible |
| Matrix Effects | Post-extraction addition | Signal suppression/enhancement ≤ 25% |
| Carryover | Injection of blank after high standard | ≤20% of LLOQ |
| Specificity | Analysis of blank matrix | No interference at retention times |
Internal standards are essential for reliable quantification, with deuterated analogs of target analytes representing the gold standard. When deuterated compounds are unavailable, structurally similar compounds or homologs may be used. Internal standards should be added at the beginning of the sample preparation process to correct for losses during extraction and variations in instrument response.
Addressing Matrix Effects in Decomposed Viscera
Matrix effects pose particular challenges in the analysis of decomposed viscera due to the complex and variable nature of the tissue matrix. These effects can cause signal suppression or enhancement, potentially compromising quantitative accuracy. Several approaches can mitigate matrix effects:
- Effective sample clean-up: Optimized extraction protocols to remove interfering compounds
- Matrix-matched calibration standards: Preparing calibration standards in processed blank matrix
- Standard addition method: Adding known amounts of analyte to aliquots of the sample
- Effective use of internal standards: Especially isotope-labeled internal standards that co-elute with target analytes
- GC×GC-MS: Improved separation of analytes from matrix interferences [21]
Method development should include specific assessment of matrix effects by comparing the response of standards in neat solvent to standards prepared in processed matrix. Significant matrix effects (typically >25% suppression or enhancement) should be addressed through method modification to ensure reliable quantification.
Analytical Workflow Visualization
The following diagram illustrates the comprehensive workflow for analysis of intoxicants in decomposed viscera using GC-MS:
GC-MS Workflow for Viscera Analysis
Research Reagent Solutions and Materials
Table 3: Essential Research Reagents and Materials for GC-MS Analysis of Intoxicants in Decomposed Viscera
| Reagent/Material | Function/Application | Technical Specifications |
|---|---|---|
| HP-5MS Capillary Column | Chromatographic separation | 30 m × 0.25 mm × 0.25 μm (5% phenyl polysiloxane) |
| Deuterated Internal Standards | Quantification control | Deuterated analogs of target analytes (e.g., Diazepam-d5, Morphine-d3) |
| BSTFA with 1% TMCS | Derivatization reagent | Silylation of polar compounds (alcohols, acids, amines) |
| Ethyl Acetate (HPLC Grade) | Extraction solvent | Low UV cutoff, low artifact formation |
| n-Hexane (HPLC Grade) | Extraction solvent | Low UV cutoff, minimal interference |
| Phosphate Buffer (pH 7.4) | Homogenization medium | Physiological pH for tissue preservation |
| Helium (99.999%) | GC Carrier gas | High purity to minimize system contamination |
| Calibration Mix | Quantitative standards | Certified reference materials at known concentrations |
| N-Methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) | Alternative derivatization | Particularly effective for steroids and cannabinoids |
| SPE Cartridges (C18) | Sample clean-up | 500 mg/6mL capacity for complex matrices |
Data Interpretation and Analytical Challenges
Compound Identification and Confirmation
In GC-MS analysis, compound identification relies on two primary parameters: retention time/index and mass spectral match. For forensic applications, a minimum of three characteristic ions should be monitored for each compound, with their relative abundances matching the reference standard within specified tolerances (typically ±20-30%). Retention time should match the reference standard within ±0.2% under the same analytical conditions. Modern data systems provide automated library searching capabilities against commercial mass spectral databases (e.g., NIST, Wiley), but manual verification is essential, particularly for compounds present at low concentrations or in complex matrices.
For decomposed viscera, additional confirmation techniques may be necessary due to matrix interferences. These include:
- Use of alternative columns with different stationary phases to confirm retention characteristics
- High-resolution mass spectrometry for exact mass measurement when available
- Chemical derivatization to produce predictable mass spectral changes
- GC×GC-MS for enhanced separation confidence [21]
Addressing Decomposition-Related Artifacts
Decomposed viscera present unique interpretive challenges due to the presence of endogenous decomposition products and potential postmortem changes to target analytes. Microbial activity, enzymatic processes, and chemical degradation can transform parent compounds into metabolites or degradation products, potentially complicating toxicological interpretation. Common issues include:
- Postmortem redistribution: Concentration differences between anatomical sites
- Postmortem production: Ethanol and other volatiles generated during decomposition
- Analyte degradation: Instability of certain compounds during storage or analysis
- Matrix enhancement/suppression: Altered ionization efficiency due to co-eluting compounds
Proper controls, including analysis of blank matrix and authentic standards prepared in matrix, are essential for distinguishing artifacts from true analytical findings. Interpretation should always consider the postmortem interval, storage conditions, and sample handling history when evaluating results.
The analysis of intoxicants in decomposed viscera using GC-MS represents a sophisticated application of analytical technology to address complex forensic questions. Through optimized sample preparation, careful method validation, and appropriate data interpretation, reliable results can be obtained even from challenging postmortem specimens. The protocols and guidelines presented in this technical guide provide a framework for conducting such analyses within the rigorous standards required for forensic applications. Continued advancement in GC-MS technology, particularly through approaches like GC×GC-MS, promises enhanced capability for addressing the analytical challenges posed by decomposed tissue matrices, ultimately supporting the accurate administration of justice through reliable scientific evidence.
In forensic science, the reliability of analytical data is paramount, as it directly influences judicial outcomes and public safety. Method validation serves as the foundational process that ensures forensic techniques and tools yield accurate, reliable, and repeatable results that are legally admissible in court [58]. This process is not merely a technical formality but an ethical and professional commitment underpinning the credibility of the entire justice system [58]. Without rigorous validation, forensic conclusions risk severe consequences, including miscarriages of justice, wrongful convictions, and loss of credibility for forensic laboratories [58].
The application of method validation is particularly critical in drug-related crimes, where the escalating incidence of such offenses demands rapid and reliable forensic screening methods [44]. Gas Chromatography-Mass Spectrometry (GC-MS) has long been the gold standard in forensic drug analysis due to its high specificity and sensitivity [44] [59]. However, traditional GC-MS methods often require extensive analysis times, hindering rapid law enforcement responses and contributing to significant case backlogs in forensic laboratories worldwide [44] [59]. This context has driven the development and implementation of rapid GC-MS technologies, which necessitate equally rigorous but efficiently executable validation protocols to ensure their reliability within judicial processes [59] [46].
Core Principles of Forensic Method Validation
Forensic validation encompasses three distinct but interconnected components: tool validation, which ensures forensic software or hardware performs as intended without altering source data; method validation, which confirms analytical procedures produce consistent outcomes across different cases and practitioners; and analysis validation, which evaluates whether interpreted data accurately reflects its true meaning and context [58]. These components collectively ensure that forensic conclusions are scientifically sound and forensically defensible.
The principles of forensic method validation are universally applicable across disciplines but hold particular significance in analytical chemistry applications like GC-MS. Core principles include [58]:
- Reproducibility: Results must be repeatable by other qualified professionals using the same method under similar conditions.
- Transparency: All procedures, software versions, logs, and chain-of-custody records must be thoroughly documented.
- Error Rate Awareness: Forensic methods should have known error rates that can be disclosed in reports and during testimony.
- Peer Review: Validation processes should be reviewed and ideally published to allow scrutiny from the broader forensic community.
- Continuous Validation: As technology evolves rapidly, tools and methods must be frequently revalidated to account for updates and new applications.
Legal standards such as the Frye and Daubert Standards require that scientific methods used in court be generally accepted in the field or demonstrably reliable, often judged by factors such as testability, error rates, and peer review [58]. Method validation provides the empirical evidence necessary to meet these legal thresholds, creating an essential bridge between scientific analysis and judicial acceptance.
Validation Frameworks and Standards
The forensic community relies on established frameworks and standards to ensure consistent validation practices across laboratories and disciplines. The Organization of Scientific Area Committees (OSAC) for Forensic Science maintains a registry of approved standards that provide guidelines for various forensic disciplines, including drug analysis [60]. As of January 2025, the OSAC Registry contained 225 standards (152 published and 73 OSAC Proposed) representing over 20 forensic science disciplines [60]. These standards create a unified framework for forensic methodology, including validation requirements.
For GC-MS applications specifically, standards often reference guidelines from organizations such as the Scientific Working Group for the Analysis of Seized Drugs (SWGDRUG) and the United Nations Office on Drugs and Crime (UNODC) [44]. The National Institute of Standards and Technology (NIST) has also developed specialized resources to address the need for standardized validation protocols for emerging technologies like rapid GC-MS [59] [46]. The NIST validation template provides laboratories with a comprehensive instruction guide tailored to two forensic applications: seized drug screening and fire debris screening [59]. This free resource includes detailed information on required materials, specific analyses to perform, and automated calculation spreadsheets, enabling laboratories to implement rigorous validation processes efficiently [59].
A critical distinction in validation terminology exists between analytical method validation and clinical qualification. While "validation" and "qualification" have sometimes been used interchangeably in scientific literature, proper usage reserves "validation" for assessing analytical method performance, and "qualification" for the evidentiary process of linking a biomarker with biological processes and clinical endpoints [61]. This distinction, though emerging from clinical contexts, underscores the importance of precise terminology in forensic validation, where analytical reliability must be established independently from its interpretive significance in casework.
Case Study: Validation of Rapid GC-MS for Seized Drug Screening
Experimental Design and Methodology
A recent study developed and optimized a rapid GC-MS method that significantly reduces total analysis time from 30 to 10 minutes while maintaining forensic reliability [44]. The methodology employed an Agilent 7890B gas chromatograph system connected to an Agilent 5977A single quadrupole mass spectrometer, equipped with a 7693 autosampler and an Agilent J&W DB-5 ms column (30 m × 0.25 mm × 0.25 μm) [44]. Helium (99.999% purity) served as the carrier gas at a fixed flow rate of 2 mL/min [44].
The key to the rapid analysis was the optimization of temperature programming and operational parameters. The rapid method utilized an initial temperature of 120°C,
ramped to 300°C at 70°C/min with a 7.43-minute hold, compared to the conventional method starting at 70°C with a slower ramp rate of 15°C/min to 300°C [44]. This optimized temperature program achieved a threefold reduction in total run time while maintaining separation efficiency necessary for confident compound identification.
The validation process assessed nine critical components: selectivity, matrix effects, precision, accuracy, range, carryover/contamination, robustness, ruggedness, and stability [46]. Single- and multi-compound test solutions of commonly encountered seized drug compounds were used to evaluate method and system performance against predetermined acceptance criteria [46].
Performance Results and Comparative Analysis
The validation study demonstrated significant improvements in analytical performance compared to conventional GC-MS methods. The systematic validation showed a limit of detection (LOD) improvement by at least 50% for key substances such as cocaine and heroin [44]. Specifically, the method achieved detection thresholds as low as 1 μg/mL for cocaine compared to 2.5 μg/mL with conventional methods [44]. This enhanced sensitivity is crucial for detecting trace amounts of controlled substances in complex forensic samples.
The method exhibited excellent repeatability and reproducibility with relative standard deviations (RSDs) less than 0.25% for stable compounds under operational conditions [44]. In precision and robustness studies, retention time and mass spectral search score % RSDs were ≤ 10%, meeting the designated acceptance criteria for forensic applications [46]. When applied to 20 real case samples from Dubai Police Forensic Labs, the rapid GC-MS method accurately identified diverse drug classes, including synthetic opioids and stimulants, with match quality scores consistently exceeding 90% across tested concentrations [44].
Table 1: Comparative Method Performance Metrics
| Performance Characteristic | Rapid GC-MS Method | Conventional GC-MS Method |
|---|---|---|
| Total Analysis Time | 10.00 minutes | 30.33 minutes |
| Cocaine LOD | 1 μg/mL | 2.5 μg/mL |
| Retention Time RSD | ≤ 10% | Similar or higher |
| Carryover/Contamination | Minimal/Controlled | Similar or higher |
| Match Quality Scores | > 90% | Typically lower |
Table 2: Validation Results for Key Compounds
| Compound | Molecular Formula | Molecular Mass (Da) | Retention Time Precision (RSD) | Spectral Match Quality |
|---|---|---|---|---|
| Cocaine | C17H21NO4 | 303.35 | < 0.25% | > 90% |
| Heroin | C21H23NO5 | 369.41 | < 0.25% | > 90% |
| Methamphetamine | C10H15N | 149.23 | < 0.25% | > 90% |
| MDMA | C11H15NO2 | 193.25 | < 0.25% | > 90% |
| Δ9-THC | C21H30O2 | 314.46 | < 0.25% | > 90% |
Despite these robust performance characteristics, the validation process appropriately identified methodological limitations, including the inability to differentiate some isomers, providing crucial context for the method's appropriate application in casework [46]. This honest assessment of limitations represents a critical component of comprehensive validation, ensuring analysts understand the boundaries of the method's capabilities.
Essential Research Reagents and Materials
The experimental work utilized carefully characterized test solutions to ensure method reliability and reproducibility. These solutions were either prepared in-house or obtained from reputable commercial sources like Cayman Chemical and Sigma-Aldrich Cerilliant [44]. Two custom "general analysis" mixtures were developed to validate the method across a broad range of forensically relevant compounds [44].
Table 3: Essential Research Reagent Solutions
| Reagent/Standard | Source | Function in Validation |
|---|---|---|
| Cocaine Standard | Sigma-Aldrich (Cerilliant) | Primary reference material for method calibration and LOD determination |
| Heroin Standard | Sigma-Aldrich (Cerilliant) | Controlled substance reference for sensitivity and specificity testing |
| Methamphetamine Standard | Commercial or in-house | Stimulant compound for separation efficiency assessment |
| MDMA Standard | Commercial or in-house | Synthetic drug representative for method validation |
| Δ9-THC Standard | Sigma-Aldrich (Cerilliant) | Cannabis compound for comprehensive drug coverage |
| Methanol (99.9%) | Sigma-Aldrich | Solvent for standard preparation and sample dilution |
| Helium Carrier Gas (99.999%) | Commercial supplier | Mobile phase for chromatographic separation |
Impact on Forensic Laboratory Efficiency and Judicial Outcomes
The implementation of properly validated rapid GC-MS methods has a substantial impact on forensic laboratory efficiency and the administration of justice. By reducing analysis time from 30 minutes to 10 minutes per sample while maintaining analytical rigor, laboratories can significantly reduce case backlogs without compromising result quality [44]. This improved throughput is particularly crucial given the "escalating incidence of drug-related crimes" and the persistent backlogs in forensic laboratories, especially in the seized drug field [44] [59].
The efficiency gains extend beyond mere speed, facilitating faster judicial processes and law enforcement responses [44]. When forensic analysis accelerates without sacrificing reliability, criminal cases can proceed more rapidly through the justice system, potentially reducing pretrial detention times and enabling more timely resolutions for victims and defendants alike [59]. This alignment between analytical efficiency and judicial efficiency represents a critical advancement in forensic science practice.
The adoption of standardized validation templates, such as those provided by NIST, further enhances these benefits by reducing the barrier to implementing new technologies [59] [46]. By providing a comprehensive validation package with detailed instructions, required materials, and automated calculation spreadsheets, these resources enable laboratories to validate new instruments in months rather than years, returning analysts to casework more quickly while ensuring the scientific integrity of their results [59]. This standardized approach to validation creates consistency across laboratories, enhancing the reliability and comparability of forensic results presented in judicial proceedings nationwide.
Method validation represents the essential bridge between analytical science and judicial reliability, ensuring that forensic methods yield accurate, reproducible, and defensible results. The case study of rapid GC-MS validation demonstrates how rigorous evaluation of sensitivity, precision, robustness, and other performance characteristics establishes the foundation for forensically sound and judicially reliable analytical methods. As forensic technologies continue to evolve, maintaining this commitment to comprehensive validation remains paramount for upholding the integrity of both forensic science and the judicial processes it serves.
The availability of standardized validation resources from organizations like NIST provides practical pathways for laboratories to implement new technologies efficiently while maintaining the rigorous standards required for judicial acceptance. By embracing these frameworks and adhering to the core principles of forensic validation, the scientific community can continue to enhance the efficiency, reliability, and ultimately the justice impact of forensic chemical analysis.
Maintaining Peak Performance: A Systematic Guide to Troubleshooting GC-MS
In gas chromatography-mass spectrometry (GC-MS) forensics research, the reliability of analytical results is paramount. Sensitivity loss—a significant reduction in instrument response to target analytes—directly impacts the detection and quantification of crucial evidence, from illicit drugs to toxicological compounds. Such a loss can lead to false negatives, inaccurate quantitation, and ultimately, a failure to detect forensically relevant substances, undermining the integrity of an investigation. This guide provides a systematic, flowchart-driven approach to diagnosing the root causes of sensitivity loss in GC-MS systems, framed within the rigorous demands of forensic science. The principles outlined ensure that methods meet the exacting standards for admissibility in legal proceedings, where the Daubert Standard emphasizes known error rates and reliable methodology [7].
The Diagnostic Flowchart
The following flowchart provides a systematic, decision-tree-based approach to diagnosing sensitivity loss in GC-MS systems. It guides the user from initial observation to the most probable root cause, categorizing problems into inlet, column, detector, and sample-related issues.
Figure 1: A systematic flowchart for diagnosing sensitivity loss in GC-MS.
Troubleshooting Quantitative Data and Symptom Patterns
Effective diagnosis requires correlating observed symptoms with quantitative performance metrics and potential causes. The following tables summarize key data to guide the troubleshooting process.
Table 1: Quantitative Symptom Patterns and Common Causes in GC-MS Sensitivity Loss
| Symptom Pattern | Key Quantitative Indicators | Most Probable Causes |
|---|---|---|
| All peaks reduced, retention times stable [62] | - Peak height/area reduction >50%- Stable retention times (±0.1 min)- Signal-to-noise ratio decrease | - Incorrect split ratio or pulse settings [62]- Autosampler syringe malfunction [62] [63]- Incorrect inlet/detector temperatures [62]- Exhausted or contaminated ion source [62] |
| All peaks reduced, retention times shifted [62] | - Peak height/area reduction- Retention time shifts >5%- Constant retention time drift | - Incorrect carrier gas flow rate [62]- Carrier gas leak [62]- Incorrect column dimensions in method [62] |
| Peak broadening with/without retention shifts [62] | - Increased peak width at half height- Loss of resolution- Tailing factor >2.0 | - Column degradation (active sites) [62]- Incorrect column installation [62]- Stationary phase damage [62] |
| Severe sensitivity loss in specific modes | - SIM sensitivity loss >1000x vs. historical data [63]- Poor LOD/LOQ in scan mode | - MS tune failure (e.g., repeller voltage spike) [62]- Incorrect SIM ions or dwell times [62]- Electron multiplier exhaustion [62] |
Table 2: Forensic GC-MS Analytical Performance Benchmarks and Impact of Sensitivity Loss
| Analytical Parameter | Typical Forensic Benchmark | Status Indicating Problem |
|---|---|---|
| Limit of Detection (LOD) | e.g., 15 ng/mL for ∆9-THC in blood [64] | LOD increases significantly over validation data |
| Limit of Quantification (LOQ) | e.g., 25 ng/mL for ∆9-THC in blood [64] | LOQ fails to meet regulatory thresholds |
| Signal-to-Noise (S/N) | Typically >10:1 for quantification [65] | S/N falls below 10:1 for target analytes |
| Linearity (R²) | >0.990 for calibration curves [64] | Consistent non-linearity or poor fit |
| Measurement Accuracy | Recovery rates of 70-140% [66] | Recovery consistently outside acceptable range |
| Precision (% RSD) | Intra- and inter-day variability ≤22% [66] | % RSD exceeds method validation criteria |
Detailed Experimental Protocols for System Diagnosis
Protocol 1: Systematic Inlet and Autosampler Inspection
A methodical check of the inlet and autosampler is critical, as these are common sources of sensitivity loss.
Leak Check and Septum Replacement:
- Pressurize the system and use an electronic leak detector to check all inlet fittings [63].
- If unavailable, apply a leak-check solution to the inlet nut, septum cap, and ferrule areas while monitoring the system pressure for drops [63].
- Replace the inlet septum if it has exceeded its recommended lifespan or shows any signs of piercing or damage. A leaking septum can cause sample loss and erratic retention times [62].
Liner Inspection and Replacement:
- Remove the inlet liner and inspect for debris, broken glass wool, or a non-volatile residue buildup. These active sites can adsorb analytes, leading to peak tailing and sensitivity loss [62].
- Replace with a clean, deactivated liner that is appropriate for the application (e.g, liner with glass wool for dirty matrices).
- Ensure the correct liner has been installed as per the method requirements [62].
Syringe and Autosampler Function Test:
- Visually inspect the autosampler syringe for bent needle, damaged plunger, or air gaps [62] [63].
- Observe a full injection cycle to confirm the syringe aspirates the correct sample volume from the vial and dispenses it smoothly into the inlet without leaking [62].
- Perform a test injection using a dye-colored solution to visually confirm the sample is being introduced into the inlet.
Method Parameter Verification:
Protocol 2: Column and Carrier Gas System Verification
Issues with the column or carrier gas can manifest as sensitivity loss, retention time shifts, and peak shape degradation.
Carrier Gas Flow Verification:
Column Installation and Dimension Check:
- Verify that the column is correctly installed in both the inlet and the MS interface. For the MS, the column tip should typically be positioned 1-2 mm from the end of the transfer line [63].
- Confirm that the physical column dimensions (length, diameter, and film thickness) match the parameters entered into the data system, as these values are critical for accurate flow and retention time calculations [62].
Column Performance Assessment:
- If column degradation is suspected (e.g., peak tailing for active compounds), trim 0.5–1 meter from the inlet side to remove the most contaminated/deactivated section [62].
- Run a column test mix containing compounds that assess efficiency and activity. Compare the results (peak symmetry, resolution, retention times) to a chromatogram obtained when the column was new or performing well [62].
Protocol 3: Mass Spectrometer Diagnostic Tuning and Evaluation
The mass spectrometer itself is a complex component where sensitivity can be lost at several points.
Tune Report Analysis:
- Examine the automated tune report for key indicators. A dramatic increase in the repeller or accelerator voltage often signals a dirty ion source that requires cleaning [62].
- A significant increase in the electron multiplier (EM) voltage to achieve a target abundance indicates a worn-out detector that may need replacement [62].
- Verify that the ionization energy is correctly set, typically 70 eV for electron ionization (EI) [62].
Detector and Mode-Specific Checks:
- For Flame Ionization Detectors (FID) and other flame-based detectors, check that the fuel gas ratios (hydrogen and air) are appropriate using a flow meter [62].
- For MS detectors in Selected Ion Monitoring (SIM) mode, verify the masses and dwell times for each ion within the SIM groups to ensure sufficient data points are being acquired across the peak [62].
- In full scan mode, check the mass range and scan speed to ensure they are appropriate for the expected peak widths and number of components [65].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for GC-MS Forensic Research and Maintenance
| Reagent/Material | Function in Forensic GC-MS | Example in Application |
|---|---|---|
| Deactivated Inlet Liners | Provides an inert surface for sample vaporization, minimizing analyte adsorption and degradation. | Used in all analyses; a liner with glass wool is often selected for dirty matrices like biological extracts [62]. |
| Derivatization Reagents (e.g., MSTFA) | Increases volatility and thermal stability of polar analytes (e.g., acids, metabolites) for GC analysis. | Essential for analyzing ∆9-THC-COOH in urine; using MSTFA allows for sensitive detection by GC-MS [64]. |
| Analyte Protectants (e.g., Gulonolactone, Sorbitol) | Compounds that mask active sites in the GC system, reducing matrix effects and improving peak shape and response. | Added to calibration standards and samples to compensate for matrix-induced enhancement in complex flavor or pesticide analysis [67]. |
| Certified Reference Materials | Provides absolute identity confirmation and enables accurate quantification against a traceable standard. | Critical for validating methods for drugs like nitazenes or cannabinoids where legal thresholds exist [68]. |
| Tuning Standards (e.g., PFTBA or DFTPP) | Allows performance verification and calibration of the mass spectrometer's mass axis, abundance, and resolution. | Used in automated tuning procedures to ensure the MS meets sensitivity and mass accuracy specifications for forensic work [62]. |
| High-Purity Solvents | Serves as the medium for sample preparation, standard dilution, and extraction to minimize background interference. | Used in liquid-liquid extraction of cannabinoids from blood and urine to achieve high recovery and clean extracts [64]. |
In gas chromatography-mass spectrometry (GC-MS) forensics research, the integrity of results is paramount. The analytical chain, however, is susceptible to specific physical degradation that manifests primarily as peak broadening and retention time shifts. These symptoms are not mere inconveniences; they directly compromise compound identification and quantification, potentially undermining the legal defensibility of data. The root causes frequently trace back to the foundational triad of the GC system: the carrier gas flow, the inlet condition, and the chromatographic column health. This guide details a systematic diagnostic and restorative approach to these issues, ensuring that data remains reliable, reproducible, and forensically sound.
Fundamental Principles: Flow, Retention, and Peak Shape
A precise understanding of the relationship between system parameters and chromatographic output is essential for effective troubleshooting.
The Role of Carrier Gas Flow
The carrier gas is the mobile phase, transporting analytes through the column. Its flow characteristics are critical for retention time reproducibility. Modern GC systems can operate in constant pressure or constant flow modes [69]. In constant flow mode, the instrument ramps the inlet pressure to maintain a consistent linear velocity of the carrier gas as the oven temperature increases and the gas viscosity changes [69] [70]. Fluctuations in flow, whether from leaks, obstructions, or incorrect instrument settings, will directly cause retention time shifts [62] [71] [70].
The average linear velocity and holdup time (tM), which is the time required for an unretained substance to traverse the column, are foundational calculations [69]. tM can be measured directly by injecting a non-retained compound like methane or butane (from a gas lighter) [69]. A symmetrical peak shape from such an injection also confirms a leak-free system with proper connections [69].
The Critical Link: Inlet Condition and Column Health
The inlet serves as the gateway for the sample, and its condition dictates the quality of the analysis from the very beginning. A contaminated or thermally degraded inlet liner, a leaking septum, or an active (deactivated) liner can cause peak tailing, broadening, and sensitivity loss for specific compounds [72] [70]. The column itself is a living component. Over time, non-volatile residues from sample matrices accumulate at the inlet end, creating active sites that adsorb analytes, leading to peak tailing and a gradual loss of chromatographic efficiency [72] [70]. This degradation is a primary cause of the combined symptom of peak broadening and retention shift.
A Systematic Diagnostic Framework
When confronting peak broadening and retention shifts, a logical, stepwise investigation is required to correctly identify the root cause. The following diagnostic workflow provides a structured path from problem observation to likely cause.
Diagnostic Workflow for Peak Abnormalities
The diagram below outlines the logical decision process for diagnosing common GC-MS issues.
Symptom-Based Investigation Guide
The table below cross-references observed chromatographic symptoms with their potential causes and immediate investigative actions. This enables rapid problem triage.
Table 1: Symptom-Based Diagnostic Guide for GC-MS Peak Issues
| Symptom Profile | Primary Suspects | Diagnostic Actions |
|---|---|---|
| All peaks broadened; retention times shifted [62] [73] | Loss of column efficiency from contamination or degradation; Incorrect carrier gas flow rate [62] [72]. | Run column test mix; Trim column inlet (0.5-1 m); Verify carrier gas flow with calibrated meter [62] [72]. |
| All peak sizes reduced; retention times shifted [62] [73] | Incorrect column dimensions entered in data system; Carrier gas flow programming error (e.g., not in constant flow mode) [62] [70]. | Check and correct column parameters (length, ID, film thickness) in data system; Confirm constant flow operation mode is selected [62]. |
| Peak tailing (all or specific peaks) [72] [70] | Active sites in inlet or column inlet; Contaminated or incorrect inlet liner; Dead volume from poor column installation [72] [70]. | Inspect and replace inlet liner; Trim column inlet (10-30 cm); Verify column is cut squarely and installed to correct depth in inlet/detector [72]. |
| Retention time shifts in initial injections after idle period [71] | System not fully equilibrated; Moisture buildup on column; Carrier gas not left flowing [71]. | Leave carrier gas flowing at low rate during idle periods; Perform several conditioning injections before running samples [71]. |
| Later-eluting peaks broadened significantly [70] | Loss of efficiency exacerbated by operating in constant pressure mode, which leads to reduced linear velocity at higher oven temperatures [70]. | Switch to constant flow mode to maintain optimal linear velocity and efficiency throughout the temperature program [70]. |
Experimental Protocols for Diagnosis and Restoration
This section provides detailed, actionable methodologies for confirming diagnoses and restoring system performance.
Protocol 1: Measuring Gas Holdup Time (tM)
Purpose: To accurately determine the holdup time (tM), a critical value for calculating retention factors and diagnosing flow-related issues [69].
Materials:
- Butane lighter or source of methane gas
- Gas-tight syringe (e.g., 5 µL for capillary systems)
- GC-MS system with installed column
Procedure:
- Ensure the GC oven is set to an isothermal temperature appropriate for your column (e.g., 40°C for a standard capillary column).
- Draw approximately 1-2 µL of vapor from the butane lighter into the syringe.
- Inject the vapor into the GC inlet as you would a liquid sample.
- Acquire data and note the retention time of the resulting peak. A sharp, symmetrical peak indicates a well-connected, leak-free system. The retention time of this peak is tM [69].
- Precaution: Butane may be retained on very thick-film columns; in such cases, methane is the preferred unretained marker [69].
Protocol 2: Evaluating Inlet and Column Inlet Condition
Purpose: To assess and remediate contamination at the column inlet, a primary cause of peak tailing and broadening.
Materials:
- GC column cutter or ceramic scal
- Magnifying glass or microscope
- New, deactivated inlet liner and septum
- Flow meter (electronic or soap-bubble)
Procedure:
- Leak Check: After cooling the system, perform a leak check at the column connections.
- Inlet Inspection: Shut off carrier gas and vent the system. Open the inlet and carefully remove the column.
- Visual Inspection: Examine the first few centimeters of the column at the inlet end. Discoloration (brown or black residue) indicates contamination [72].
- Column Trimming: Using a proper column cutter, trim 10-30 cm from the inlet end. Inspect the cut with a magnifying glass to ensure it is square and clean, with no jagged edges [72] [70].
- Liner Replacement: Replace the inlet liner with a new, deactivated one appropriate for your application (e.g., packed with deactivated wool for dirty samples).
- Reinstall & Test: Reinstall the column according to the manufacturer's specified depths. After pressurizing, use a flow meter to verify the column flow rate. Finally, run a column test mix and compare peak shapes and efficiency to the column's original quality control report [72].
Protocol 3: Verifying Carrier Gas Flow Integrity
Purpose: To independently confirm the accuracy of the electronically reported carrier gas flow rate, ruling out sensor drift or calculation errors.
Materials:
- Calibrated electronic flow meter or soap-bubble flow meter
Procedure:
- Set Conditions: Ensure the GC is at a stable, isothermal temperature (e.g., 50°C) and operating in constant flow mode.
- Measure Outlet Flow:
- For detectors like FID: Attach the flow meter to the column outlet (after detaching it from the detector) and measure the volumetric flow rate [69].
- Critical Safety Note: The column outlet must NOT be connected to a live detector (especially an FID or MS) during this measurement.
- Compare Values: Compare the measured flow rate to the value set in the method and calculated by the data system. A significant discrepancy suggests incorrect column parameters are entered, or there is a leak or obstruction [62].
The Scientist's Toolkit: Essential Research Reagent Solutions
Maintaining a consistent inventory of high-quality consumables is the first line of defense against chromatographic degradation.
Table 2: Essential Consumables for Robust GC-MS Operation
| Item | Function & Importance |
|---|---|
| Ultra-High Purity (UHP) Carrier Gas with moisture/hydrocarbon traps | Prevents stationary phase degradation and baseline noise caused by impurities, which is critical for trace-level and MS analysis [72]. |
| Deactivated Inlet Liners (various geometries: straight, tapered, gooseneck) | Provides an inert vaporization chamber. The correct geometry and packing (e.g., deactivated wool) minimize discrimination and decomposition for different sample types [72] [73]. |
| High-Temperature Septa | Maintains inlet seal during injection and under high temperature. A leaking septum introduces oxygen and causes retention time shifts [72] [71]. |
| Guard Columns/Retention Gaps | Short (0.5-5 m) lengths of deactivated tubing connected before the analytical column. They trap non-volatile residues, protecting the expensive analytical column [72]. |
| Certified Column Cutter | Ensures a clean, square cut with no debris to minimize dead volume when installing a column or after trimming [70]. |
| Calibrated Electronic Flow Meter | Provides an independent, reliable measurement of carrier and detector gas flows for system verification and troubleshooting [69] [62]. |
| Performance Test Mix | A solution of specific analytes used to benchmark system performance, including efficiency, peak symmetry, and retention time stability [72]. |
In GC-MS forensics, where data integrity is non-negotiable, a reactive approach to troubleshooting is insufficient. Peak broadening and retention time shifts are clear indicators of underlying systemic issues related to carrier gas management, inlet maintenance, and column health. By adopting the proactive, systematic diagnostic and maintenance strategies outlined in this guide—centered on regular verification of flow parameters, scheduled inspection of consumables, and strategic column trimming—researchers can ensure their instruments deliver the high-fidelity data required for definitive compound identification and legally defensible results. A well-maintained GC-MS system is not just a tool but the foundation of reliable forensic science.
In Gas Chromatography-Mass Spectrometry (GC-MS) forensics research, the integrity of analytical data is paramount. The ion source and detector are the core components responsible for generating and measuring the signal that identifies and quantifies chemical substances. A sudden drop in mass spectrometry (MS) signal is a frequently encountered issue that can severely compromise the analysis of seized drugs, toxicological samples, and other forensic evidence. Such signal loss can lead to false negatives, inaccurate quantification, and ultimately, a breakdown in the judicial process. This guide, framed within the principles of GC-MS for forensics, provides an in-depth examination of the causes and solutions for signal drop, with a focused look at maintaining the ion source and detector. A systematic approach to maintenance and troubleshooting is not merely a technical exercise but a fundamental requirement for ensuring the reliability and admissibility of forensic evidence.
Understanding Signal Drop: Causes and Initial Diagnosis
A sudden loss of sensitivity is one of the most disconcerting problems in a forensic lab. For instance, a researcher reported a sensitivity drop to 10% of previous levels within a week while running LC-MS/MS methods for nerve agents, with cleaning providing only a temporary, minor improvement [74]. This underscores that while a dirty source is a common culprit, it is not the only one.
Systematic Troubleshooting Workflow
Before disassembling the instrument, follow a logical troubleshooting workflow to isolate the problem. The initial step is to determine whether the issue originates from the sample, the liquid chromatography (LC) system, or the mass spectrometer itself [75].
The following diagram outlines a systematic diagnostic approach to isolate the cause of signal loss, starting from the simplest checks to more complex instrument diagnostics:
A critical real-world example illustrates this process: after a three-week instrument hiatus, a researcher observed a complete loss of signal. A direct infusion of standard, bypassing the LC, confirmed the MS source was functioning. Reconnecting the LC flow revealed a reciprocating signal artifact. The root cause was an air pocket in the LC pump that prevented proper solvation of analytes on the column, which was resolved by manually priming the pump [75]. This case highlights that the problem may not be the MS itself, but the LC delivery system.
Ion Source Maintenance and Cleaning
The ion source is where sample molecules are ionized, a process critical to the generation of signal. Contamination here is a primary cause of sensitivity loss.
Detailed Experimental Protocol: Ion Source Cleaning
The following protocol is adapted from standard procedures for cleaning an Agilent GC-MS ion source [76]. Always consult your specific manufacturer's manual before beginning.
- Objective: To restore ionization efficiency and signal sensitivity by removing non-volatile residues and contaminants that accumulate during normal operation.
- Principle: Abrasive cleaning with aluminum oxide slurry mechanically removes tenacious deposits, followed by sequential sonication in solvents to dissolve and wash away organic and inorganic contaminants.
- Materials and Reagents:
- Personal Protective Equipment (PPE): Nitrile or lint-free gloves, safety glasses.
- Abrasive Slurry: Aluminum oxide powder of 1-micron or finer grade, suspended in reagent-grade methanol or isopropanol.
- Solvents: Sequence of reagent-grade or higher purity solvents: Methanol, Acetone, and Dichloromethane (or a suitable alternative).
- Tools: Dedicated soft-bristle brushes (e.g., small test tube brush), compressed air or nitrogen gas source, low-power Dremel-type tool (optional, for slurry polishing), ultrasonic bath, set of non-metallic forceps.
- Methodology:
- Disassembly: Power down the mass spectrometer and vent the vacuum according to the manufacturer's procedure. Carefully remove the ion source assembly. Disassemble the individual components (e.g., repeller, focus plates, insulator) as per the instrument manual.
- Initial Inspection: Visually inspect all parts for discoloration, pitting, or thick, crusty deposits.
- Abrasive Cleaning (if heavily contaminated): Apply a small amount of aluminum oxide slurry to a soft brush or a Dremel tool with a felt polishing tip. Gently polish all metallic surfaces of the ion source components, taking care to avoid sharp edges and electrical contact points. Do not use a high-power Dremel, as it can rapidly ruin stainless-steel parts [76].
- Sonication:
- Place the components in a glass beaker and submerge in methanol.
- Sonicate for 15 minutes.
- Discard the solvent and repeat the sonication with fresh methanol.
- Perform a final sonication in a volatile solvent like acetone or dichloromethane for 15 minutes to promote rapid drying.
- Drying and Reassembly: Use a stream of clean, dry compressed air or nitrogen to thoroughly dry all parts. Ensure all solvent residue is removed. While handling parts only with clean, lint-free gloves to prevent finger oils from contaminating the surfaces, reassemble the ion source [76].
- Reinstallation and Verification: Reinstall the source into the mass spectrometer, pump down to vacuum, and allow the system to equilibrate. Perform a mass spectrometer tune and compare the results, particularly the required electron multiplier voltage and the relative abundances of tuning ions, to a baseline tune from when the source was known to be clean.
Detector Maintenance and Performance Assessment
The detector, typically an electron multiplier (EM), amplifies the small ion current into a measurable signal. Its degradation is a common source of gradual signal loss.
Electron Multiplier Function and Failure Modes
Electron multipliers function by creating a cascade of electrons when an ion strikes the first dynode. Two common designs are continuous dynode and discrete dynode multipliers. Discrete dynode designs often provide increased ion detection efficiency and longer lifetimes [76]. Over time and with use, the coating on the dynodes is depleted, reducing the detector's ability to amplify the signal. This forces the instrument to apply a higher voltage to the EM to maintain the same signal output.
Quantitative Assessment and Replacement Guide
The primary quantitative indicator of EM health is the voltage required to achieve a specified sensitivity during the instrument's auto-tune procedure. A steadily increasing voltage indicates the EM is nearing the end of its useful life.
Table 1: Performance Metrics and Maintenance Schedule for Key GC-MS Components
| Component | Key Performance Metric | Typical Maintenance Action | Replacement Indicator |
|---|---|---|---|
| Electron Multiplier | EM Voltage [76] | Monitor voltage during monthly tune | Voltage is 500-1000 V above new baseline or near maximum specification |
| Ion Source | Signal intensity of tuning ions; Required EM voltage [76] | Clean every 1-3 months or as needed [77] | >50% sensitivity loss; severe contamination not resolved by cleaning |
| Filaments | Tune stability; emission current | Replace in pairs; keep spare | Failure to pass tune; no emission current |
| Vacuum Pump | Foreline and high vacuum pressure readings; pump downtime [77] | Change rough pump oil every 6-12 months [76] | Inability to reach or maintain operating vacuum |
Replacement is a key maintenance skill. For certain models, designs allow the complete mass filter assembly to be removed to access the channel electron multiplier, which can then be replaced without the need for special tools or spot welding equipment [78]. For quadrupole systems, detectors like the channel electron multiplier SEM-6M are designed to withstand multiple cycles between vacuum and atmosphere without performance degradation, a valuable feature for maintenance [79].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful maintenance relies on having the correct supplies on hand. The following table details essential items for ion source and detector care.
Table 2: Essential Materials for Ion Source and Detector Maintenance
| Item | Function | Specific Use Case |
|---|---|---|
| Aluminum Oxide Slurry (1µm) | Abrasive cleaning of metal source parts | Removing tenacious, non-volatile deposits from repeller and ion volume [76] |
| Reagent-Grade Solvents (Methanol, Acetone) | Solubilizing and removing organic contaminants | Sonication sequence for cleaned ion source parts; final rinse with volatile solvent for drying [76] |
| Graphite/Vespel Ferrules | Creating a leak-free seal at MS interface | Withstands higher sealing force and temperature compared to pure graphite; requires re-tightening after heating [76] |
| Discrete Dynode Electron Multiplier | High-gain ion signal detection | Replacement detector offering longer lifetime and robust performance for Agilent systems [76] |
| Inland 45 Pump Oil | Lubrication and sealing for rough pumps | Recommended for lower vapor pressure and better vacuum performance; reduces risk of oil backstreaming [76] |
| Pre-aligned Filament Assemblies | Electron generation for EI source | Easy replacement without need for manual alignment; spare set should be kept on hand [78] |
Integrated Preventive Maintenance Schedule
A proactive approach is crucial for minimizing unplanned downtime in a high-throughput forensic lab. The following diagram synthesizes the maintenance of the ion source, detector, and related systems into a coherent timeline.
This schedule should be integrated with GC-specific maintenance, such as replacing the inlet liner every 100-200 injections and the septum every 50-100 injections [80]. Furthermore, preventive maintenance includes checking all gas filters and the EPC pressure zero every month [80].
In forensic GC-MS research, the data produced is only as reliable as the instrument that generates it. A sudden signal drop is a serious event, but it can be systematically diagnosed and resolved through a thorough understanding of the ion source and detector. Adherence to detailed cleaning protocols, vigilant monitoring of performance metrics like EM voltage, and the implementation of a rigorous preventive maintenance schedule are non-negotiable best practices. By mastering these procedures, researchers and lab professionals can ensure their instruments operate at peak performance, thereby upholding the stringent requirements for accuracy, reliability, and validity in forensic science.
Preventative Maintenance and Supply Selection for Future-Proofing Analyses
In gas chromatography-mass spectrometry (GC-MS) forensics research, the integrity of analytical results is paramount. These results underpin critical decisions in legal proceedings and pharmaceutical development, where accuracy and reproducibility are non-negotiable. This guide establishes a framework for future-proofing GC-MS analyses through a disciplined, principled approach to preventative maintenance and the strategic selection of consumables. By adhering to core principles of proactive instrument care and chromatographic science, researchers can safeguard their data against common pitfalls, minimize operational downtime, and ensure that their methods remain robust and defensible for years to come. A foundational understanding of the GC-MS workflow is essential for diagnosing issues and planning effective maintenance; the following diagram outlines the core process and its key components.
Foundational Principles of GC-MS Maintenance
Preventative maintenance is a strategic practice, not a reactive chore. Its implementation is guided by several key principles that directly impact data quality in a forensic context.
The Principle of Proactive Consistency
Consistency in Results: Regular maintenance is the primary strategy for minimizing variations in analytical results, which is a cornerstone of forensic method validation [81]. A well-maintained instrument delivers precise and reliable data, ensuring that results are reproducible over time and across different batches of casework samples. Longer Instrument Lifespan: Proper care significantly extends the operational life of a GC-MS system, protecting the laboratory's investment and ensuring the long-term continuity of analytical methods, which is critical for re-analysis in cold cases [81].
The Principle of Contamination Control
Cost Savings: A preventive maintenance regimen is more cost-effective than major reactive repairs. Identifying and addressing minor issues, such as a small leak or a slightly contaminated source, prevents them from escalating into catastrophic failures that require expensive parts and service [81]. Safety Assurance: Ensuring the integrity of the GC-MS system minimizes the risk of accidents, such as gas leaks or electrical faults, thereby protecting laboratory personnel and the physical evidence being analyzed [81].
A Systematic Preventative Maintenance Protocol
A structured, documented maintenance schedule is the practical implementation of preventative principles. The following protocols are essential for maintaining instrument fidelity.
Daily and Weekly Maintenance Activities
- Daily Tuning and Performance Verification: Perform a daily tune and mass calibration using a standard compound like perfluorotributylamine (PFTBA) [82] [83]. This is an invaluable tool for monitoring the health of the ion source and mass analyzer. Examine the tune report for indicators of emerging issues, such as changes in sensitivity, resolution, or the appearance of air and water peaks (m/z 28, 32, 18) suggesting a leak.
- Gas Supply and Pressure Checks: Daily, verify carrier gas pressures and flows. Check for leaks, particularly at the inlet and column connections. Ensure that gas traps (oxygen, moisture, hydrocarbon) are functioning and have not been exhausted [83].
- Weekly Vacuum System and Rough Pump Care: On a weekly basis, inspect the roughing pump oil level and color. Dark, cloudy oil indicates contamination and should be replaced. "Ballast" the pump to expel dissolved volatiles, which optimizes pumping efficiency and instrument sensitivity [83].
- Weekly Data Backup and Disk Maintenance: Back up all acquisition methods, quantification databases, and sample data on a regularly scheduled basis [82]. Perform disk maintenance tasks to ensure the control computer operates smoothly.
Monthly and Quarterly Maintenance Procedures
- Ion Source Cleaning: Clean the ion source every 1-3 months, or as indicated by a loss of sensitivity in the daily tune or the presence of excessive background noise. Autotune data can help diagnose hardware status before disassembling the source [82].
- Inlet and Injection Port Maintenance: Monthly, replace the inlet septum and inspect the inlet liner. Clean or replace the liner if visible contamination is present. A dirty liner is a common source of peak tailing, adsorption, and analyte degradation [82].
- Column Maintenance: Trim the column by cutting 10-15 cm from the injector end to remove non-volatile residues. If performance does not improve, replace the column. When installing a new column, condition it properly by holding it at its maximum temperature for the manufacturer's recommended time [81] [83].
- Autosampler Maintenance: For liquid autosamplers, headspace samplers, and purge & trap systems, perform monthly cleaning of the syringe, needle, and associated seals to prevent cross-contamination and ensure injection volume accuracy [82].
- Full Scan Instrument Blanks: If your laboratory primarily operates in Selected Ion Monitoring (SIM) mode, run a monthly instrument blank in Full Scan (FS) mode. This helps identify non-target contamination that might otherwise go undetected and interfere with analyses [82].
Comprehensive Maintenance Schedule
Table 1: GC-MS Preventative Maintenance Schedule and Objectives
| Frequency | Maintenance Task | Key Objective | Forensic Data Impact |
|---|---|---|---|
| Daily | PFTBA Autotune & Evaluation [82] | Verify mass calibration, sensitivity, and resolution. | Ensures accurate compound identification and quantification. |
| Daily | Gas Supply/Pressure Check [81] | Maintain consistent carrier gas flow. | Prevents retention time shifts crucial for comparison. |
| Weekly | Rough Pump Inspection/Oil Change [83] | Maintain high vacuum for optimal sensitivity. | Prevents signal loss for trace-level analytes (e.g., drug metabolites). |
| Monthly | Ion Source Cleaning [82] | Remove contamination causing signal noise/loss. | Restores sensitivity for low-abundance ions; reduces spectral noise. |
| Monthly | Inlet Liner/Septa Replacement [82] | Prevent sample adsorption and decomposition. | Reduces peak tailing and ghost peaks; improves quantitative accuracy. |
| Quarterly | Column Trim/Inspection [81] | Remove non-volatile residues from column head. | Improves peak shape and resolution of target compounds. |
| As Needed | Control Charting & Blank Analysis [82] | Monitor long-term performance and detect contamination. | Provides objective evidence of method stability and data integrity. |
Strategic Selection of GC-MS Supplies and Reagents
The selection of consumables is not a mere procurement task but a critical analytical parameter. The right choices directly enhance method robustness, sensitivity, and longevity.
GC Column Selection for Forensic Applications
Choosing the correct GC column is the first and most critical step in method development, as the stationary phase directly governs the separation factor (α), which has the greatest impact on resolution [84].
- Stationary Phase Polarity and Selectivity: Select a phase with polarity complementary to your target analytes. For general forensic screening (e.g., drugs of abuse), a 5% diphenyl/95% dimethyl polysiloxane phase (e.g., Rxi-5ms, HP-5) is a versatile starting point due to its medium polarity and thermal stability [84]. For more specific applications, such as analyzing acidic drugs or metabolites, a cyanopropylphenyl phase (e.g., Rtx-1701) offers greater selectivity [84].
- Column Dimensions: For GC-MS, narrower inner diameters (e.g., 0.25 mm) provide higher separation efficiency, while shorter lengths (e.g., 15-30 m) yield faster analysis times. Always use MS-designated columns, which are certified for low bleed to minimize background noise in the mass spectrometer [83].
- The Scientist's Toolkit: The following table details key consumables and their function in ensuring reliable GC-MS operation.
Table 2: Research Reagent Solutions for GC-MS Analysis
| Supply/Reagent | Function | Selection & Optimization Guidance |
|---|---|---|
| GC Column | Separates component mixtures in time. | Choose a low-bleed "MS" phase [83]. Select stationary phase and dimensions based on application [84]. |
| Carrier Gas | Mobile phase transporting vaporized sample. | Use high-purity (≥99.999%) gas. Employ high-capacity oxygen/moisture traps [83]. Consider hydrogen for faster analysis. |
| Tuning Standard | Calibrates mass axis and optimizes ion source voltages. | PFTBA is industry standard [83]. Use for daily performance verification and diagnostic. |
| Inlet Liner | Vaporizes liquid sample in a hot, inert environment. | Use deactivated, single-gooseneck liners. Select liner volume and packing based on sample volatility and cleanliness. |
| Mass Calibration Standard | Provides ions across a broad mass range for accurate mass assignment. | Contains known compounds (e.g., FC-43). Used less frequently than tuning standard for high-accuracy calibration. |
| Vespel/Graphite Ferrules | Creates a vacuum-tight seal between column and inlet/detector. | Prevents oxygen permeation into the system, which degrades the column and increases bleed [83]. |
Optimizing MS Performance through Strategic Tuning
Moving beyond routine autotuning can yield significant gains in sensitivity and specificity.
- Electron Energy Optimization: While 70 eV is standard for library-compatible spectra, reducing the electron energy can enhance the molecular ion abundance, aiding in the identification of unknown compounds. Increasing the energy may improve the abundance of specific fragment ions for more sensitive SIM methods [83].
- Ion Source Voltage Ramping: Manually optimize voltages for the repeller, lens, and other ion source components by monitoring the abundance of ions close to your target masses. This "target tuning" can provide a significant sensitivity boost over generic autotune values [83].
- Mass Analyzer Tuning: For quadrupole systems, the balance between resolution and sensitivity can be adjusted. Increasing the DC voltage improves resolution but reduces sensitivity, while lowering the DC voltage has the opposite effect. This can be tuned to separate co-eluting isobaric interferences [83].
Troubleshooting Common GC-MS Issues
Even with rigorous maintenance, issues can arise. A systematic approach to troubleshooting, based on chromatographic symptoms, is key to rapid resolution.
Table 3: GC-MS Troubleshooting Guide for Common Analytical Issues
| Symptom | Potential Causes | Diagnostic & Corrective Experiments |
|---|---|---|
| Loss of Sensitivity | 1. Contaminated ion source [82].2. Exhausted gas traps or contaminated carrier gas [83].3. Active sites in inlet/column [82]. | 1. Diagnostic: Check daily tune report for decreased PFTBA ion abundance.Protocol: Clean or replace ion source.2. Diagnostic: Check for high m/z 28, 32 in tune; replace traps.Protocol: Install new oxygen/moisture traps.3. Diagnostic: Observe peak tailing for active compounds (e.g., alcohols, amines).Protocol: Trim column, replace inlet liner, re-silylate column. |
| Unstable Retention Times | 1. Carrier gas leak or flow instability [81].2. Faulty pressure/flow control module.3. Column not properly secured in oven. | 1. Diagnostic: Perform leak check; listen for hissing, use leak detector fluid.Protocol: Tighten fittings, replace ferrules/septa.2. Diagnostic: Monitor actual column flow vs. setpoint.Protocol: Require service for electronic pressure control (EPC) module. |
| High Background/Column Bleed | 1. Column exceeded temperature limit [83].2. Oxygen degradation of stationary phase [83].3. Contaminated inlet. | 1. Diagnostic: Look for ions 207, 281 in background spectra [83].Protocol: Condition column at or below max temp; replace if bleed persists.2. Diagnostic: Confirm with high m/z 32 in background.Protocol: Ensure gas traps are fresh; check system for leaks. |
| Ghost Peaks/Contamination | 1. Contaminated inlet liner, syringe, or autosampler [82].2. Septa bleed.3. Contaminated solvent or reagents. | 1. Diagnostic: Run a solvent blank; peaks still present.Protocol: Replace liner, clean syringe, run autosampler wash protocols.2. Diagnostic: Peaks present in blank and all samples.Protocol: Use high-temperature, low-bleed septa; replace regularly. |
Future-proofing GC-MS analyses in a forensic research environment demands a holistic strategy that intertwines disciplined preventative maintenance with intelligent supply selection. This guide has detailed the protocols for maintaining instrument integrity and the rationale behind selecting consumables that enhance analytical performance. By embracing the principles of proactive consistency, rigorous documentation, and strategic optimization, scientists and researchers can build a foundation of unwavering data integrity. This commitment to quality ensures that their analytical results are not only scientifically sound but also legally defensible, standing the test of time in the demanding field of forensic science.
Beyond Traditional GC-MS: Validation, Legal Admissibility, and Advanced Techniques
For forensic researchers and drug development professionals, the analytical precision of a technique like Gas Chromatography-Mass Spectrometry (GC-MS) is only one part of the evidentiary equation. The other, equally critical, part is ensuring that the resulting data and expert testimony meet the legal standards for admissibility in court. The judicial system functions as a gatekeeper for scientific evidence, and the pathways through which evidence is admitted are primarily defined by the Daubert Standard, the Frye Standard, and Federal Rule of Evidence 702. Understanding these frameworks is not merely a legal formality; it is a fundamental aspect of rigorous scientific practice in forensics and pharmacology. This guide provides an in-depth technical examination of these legal standards, framing them within the context of GC-MS forensic research to equip scientists with the knowledge necessary to ensure their work withstands legal scrutiny.
Core Legal Frameworks for Scientific Evidence
The Frye Standard: General Acceptance
The Frye Standard, originating from the 1923 case Frye v. United States, is the historical cornerstone for the admissibility of scientific evidence in U.S. courts [85]. The test is succinct: expert testimony is admissible only if the scientific technique on which it is based is "generally accepted" as reliable in the relevant scientific community [86] [87]. In Frye, the court affirmed the exclusion of testimony regarding a systolic blood pressure deception test, the forerunner of the polygraph, because it has not "gained general acceptance in the particular field in which it belongs" [87].
- Core Principle: The central inquiry is whether the principle or technique has achieved widespread acceptance within its field. The focus is not on the ultimate conclusions of the expert but on the general acceptance of the methodology used [87].
- Application: The standard is typically applied to novel scientific techniques. A Frye hearing, which is often narrower than a Daubert hearing, focuses almost exclusively on this single factor of general acceptance [87].
- Current Status: While the Frye standard was superseded in federal courts by the Daubert standard, it remains the governing law in several state courts, including California, Illinois, New York, and Pennsylvania [88] [86].
The Daubert Standard and the "Daubert Trilogy"
In the 1993 case Daubert v. Merrell Dow Pharmaceuticals, Inc., the United States Supreme Court established a new standard for federal courts, holding that the Federal Rules of Evidence, specifically Rule 702, had superseded the Frye standard [88]. The Court assigned trial judges a "gatekeeping" role, requiring them to ensure that all expert testimony is not only relevant but also reliable [88] [89]. The Daubert standard was further refined by two subsequent Supreme Court cases, collectively known as the "Daubert Trilogy":
- Daubert v. Merrell Dow Pharmaceuticals, Inc. (1993): Established the judge's gatekeeping role and provided a non-exclusive checklist of factors to assess the reliability of scientific testimony [88] [89].
- General Electric Co. v. Joiner (1997): Held that an appellate court should review a trial judge's decision to admit or exclude expert testimony under an "abuse of discretion" standard. It also emphasized that a court may exclude an opinion if there is "too great an analytical gap between the data and the opinion proffered" [88] [89].
- Kumho Tire Co. v. Carmichael (1999): Expanded the judge's gatekeeping obligation described in Daubert to apply to all expert testimony, including that based on "technical" and "other specialized" knowledge, not just scientific testimony [88] [90].
The Daubert Court provided five illustrative factors to guide judges in assessing reliability [88] [89]:
- Whether the theory or technique can be (and has been) tested: The scientific method is grounded in falsifiability, refutability, and testability.
- Whether the theory or technique has been subjected to peer review and publication: Peer review is a component of good science, helping to ensure that flaws in methodology are identified.
- The known or potential error rate: The court should consider the techniques' error rate and the existence and maintenance of standards controlling its operation.
- The existence and maintenance of standards controlling the technique's operation: The court should consider the techniques' error rate and the existence and maintenance of standards controlling its operation.
- Whether the theory or technique has gained general acceptance in the relevant scientific community: The Frye standard was incorporated as one factor among several in the Daubert analysis.
Federal Rule of Evidence 702: Codifying the Standard
Federal Rule of Evidence 702 codifies the principles of the Daubert trilogy. It was amended in 2000 to reflect these cases and was most recently amended in 2023 to clarify and emphasize the judge's gatekeeping responsibilities [91] [92]. The current rule states:
A witness who is qualified as an expert by knowledge, skill, experience, training, or education may testify in the form of an opinion or otherwise if the proponent demonstrates to the court that it is more likely than not that [91]:
- (a) the expert’s scientific, technical, or other specialized knowledge will help the trier of fact to understand the evidence or to determine a fact in issue;
- (b) the testimony is based on sufficient facts or data;
- (c) the testimony is the product of reliable principles and methods; and
- (d) the expert’s opinion reflects a reliable application of the principles and methods to the facts of the case.
The 2023 amendment is particularly significant. It replaced the phrase "the expert has reliably applied" with "the expert’s opinion reflects a reliable application" to stress that the proponent must prove admissibility by a preponderance of the evidence and that the court must exclude expert testimony that goes beyond what the expert's basis and methodology can reliably support [92].
Comparative Analysis: Daubert vs. Frye
The differences between the Daubert and Frye standards have practical implications for how scientific evidence is evaluated and presented.
Table 1: Key Differences Between the Daubert and Frye Standards
| Feature | Daubert Standard | Frye Standard |
|---|---|---|
| Governing Question | Is the testimony based on reliable methodology and is it relevant? [89] | Is the methodology generally accepted in the relevant scientific community? [87] |
| Scope | Applies to all expert testimony (scientific, technical, specialized knowledge) [89] [90] | Primarily applied to novel scientific techniques [87] |
| Role of the Judge | Active "gatekeeper" who assesses the reliability of the methodology [88] | Assesses whether the methodology is generally accepted; more deferential to the scientific community [87] |
| Focus of Inquiry | Methodology and its reliable application to the facts [89] | General acceptance of the methodology itself [87] |
| Flexibility | Flexible, multi-factor test [88] | Single, rigid test [85] |
Applying Legal Standards to GC-MS Forensic Research
GC-MS Fundamentals and Methodology
Gas Chromatography-Mass Spectrometry (GC-MS) is a combined analytical technique that separates chemical mixtures (gas chromatography) and identifies the individual components based on their mass (mass spectrometry) [93]. Its operation can be broken down into two core stages:
- Gas Chromatography (GC): A liquid sample is vaporized and carried by an inert gas (e.g., helium or hydrogen) through a coated column [93]. Different compounds in the sample interact with the column coating to varying degrees, causing them to elute at different retention times, thereby separating them.
- Mass Spectrometry (MS): As the separated compounds elute from the GC column, they enter the mass spectrometer. They are first ionized, commonly by Electron Ionization (EI), which fragments the molecules into characteristic ions [93]. These ions are then separated by a mass analyzer (e.g., a quadrupole) based on their mass-to-charge ratio (m/z). A detector then produces a mass spectrum, which is a unique "fingerprint" of the compound that can be used for identification and quantification [93].
Establishing GC-MS Reliability Under Daubert
For a forensic expert presenting GC-MS data, each step of the analysis must be demonstrably reliable to satisfy Rule 702 and the Daubert factors.
Table 2: Addressing Daubert Factors in GC-MS Analysis
| Daubert Factor | Application to GC-MS Analysis | Supporting Documentation & Protocols |
|---|---|---|
| Testing & Falsifiability | The fundamental principles of GC-MS are testable. Protocols can be designed to separate and identify known and unknown compounds. | Validation studies; method development and optimization data; calibration curves. |
| Peer Review & Publication | The GC-MS technique is extensively documented in thousands of peer-reviewed scientific journals and analytical chemistry textbooks. | Citations to foundational and application-specific literature; references to standard analytical method compendia. |
| Known Error Rate | The technique has established rates of false positives/negatives. Uncertainty can be quantified via validation studies. | Validation data showing specificity, accuracy, precision, limits of detection/quantification; proficiency test results. |
| Existence of Standards | Strict standards and controls govern instrument operation, method validation, and sample handling. | Standard Operating Procedures (SOPs); ISO/IEC 17025 accreditation [60]; OSAC registry standards [60]; quality control/quality assurance (QC/QA) records. |
| General Acceptance | GC-MS is universally accepted in analytical chemistry, toxicology, and forensic science for drug identification, toxicology, and trace evidence analysis. | Judicial notice of acceptance; expert testimony on its widespread use; its inclusion in OSAC standards [60]. |
The Scientist's Toolkit: Essential GC-MS Reagents and Materials
A reliable GC-MS analysis depends on high-purity materials and standardized reagents. The following table details key components of a GC-MS system and their critical functions.
Table 3: Key Research Reagent Solutions and Materials for GC-MS Analysis
| Item | Function in GC-MS Analysis |
|---|---|
| Carrier Gas (e.g., Helium, Hydrogen) | The mobile phase that transports the vaporized sample through the GC column. Must be high-purity to avoid contamination and baseline noise. |
| GC Capillary Column | A long, thin tube coated with a stationary phase where the chemical separation of the sample mixture occurs based on compound polarity and volatility. |
| Derivatization Reagents | Chemical compounds used to treat samples to improve volatility, stability, or chromatographic behavior of analytes that are otherwise difficult to analyze. |
| Certified Reference Standards | Pure, certified materials of known concentration and identity used to calibrate the instrument, validate methods, and positively identify unknown analytes. |
| Calibration Solutions | A series of solutions with known concentrations of target analytes, used to construct a calibration curve for quantitative analysis. |
| Quality Control (QC) Samples | Samples with known properties analyzed alongside unknown samples to monitor the instrument's performance and ensure the validity of the analytical run. |
| Solvents (HPLC/Grade) | High-purity solvents used to prepare samples, standards, and for system rinsing. Purity is critical to prevent introducing interfering contaminants. |
Experimental Protocol for a Forensically Defensible GC-MS Analysis
To ensure admissibility under legal standards, a GC-MS analysis for forensic purposes must follow a rigorous, documented protocol. The following workflow outlines the critical phases, from sample receipt to data interpretation and reporting.
Figure 1: GC-MS Forensic Analysis Workflow. This diagram outlines the sequential phases of a forensically defensible analytical process.
Phase 1: Sample Receipt and Chain of Custody
- Objective: To ensure the integrity and traceability of the evidence from receipt to disposal.
- Protocol: Upon receipt, document the sample condition, unique identifier, date, and time. Maintain a continuous chain of custody log that records every individual who handles the evidence. Store the sample in a secure environment to prevent contamination or degradation.
Phase 2: Sample Preparation
- Objective: To extract and prepare the analyte of interest for introduction into the GC-MS system.
- Protocol: Weigh a representative sub-sample. Perform a solid-liquid or liquid-liquid extraction using appropriate solvents (e.g., methanol, ethyl acetate) to isolate the target compounds. If analyzing for drugs or metabolites that contain polar functional groups (e.g., acids, alcohols, amines), a derivatization step is necessary. A common protocol is to react the extract with N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) to form trimethylsilyl derivatives, which improve volatility and chromatographic performance.
Phase 3: Instrument Calibration and System Suitability
- Objective: To verify that the GC-MS system is operating within specified parameters and is capable of providing valid data.
- Protocol:
- Tune the Mass Spectrometer: Use a standard like perfluorotributylamine (PFTBA) to calibrate the mass axis and ensure the instrument meets manufacturer specifications for resolution and sensitivity.
- Run Calibration Standards: Analyze a series of at least five calibration standards containing the target analytes at known concentrations (e.g., 1 ng/µL, 10 ng/µL, 100 ng/µL, 1000 ng/µL). The resulting calibration curve should have a correlation coefficient (R²) of ≥ 0.99.
- System Suitability Test: Analyze a quality control (QC) sample at a mid-range concentration. The retention times and peak areas of the QC sample must be within a pre-defined percentage (e.g., ±5%) of the expected values from the calibration curve.
Phase 4: Data Acquisition
- Objective: To generate chromatographic and mass spectral data for the prepared sample.
- Protocol: Inject a precise volume (e.g., 1 µL) of the prepared sample extract into the GC-MS system using an autosampler. The GC method should include a temperature ramp optimized to separate the target compounds. The mass spectrometer should be operated in full scan mode (e.g., m/z 40-550) for qualitative identification or in Selected Ion Monitoring (SIM) mode for enhanced quantitative sensitivity.
Phase 5: Data Interpretation and Quality Review
- Objective: To identify and quantify the target analytes and review all data for quality and consistency.
- Protocol:
- Compound Identification: A positive identification requires two independent data points:
- Retention Time Match: The sample analyte's retention time must match the calibration standard's within a narrow window (e.g., ±0.1 minutes).
- Mass Spectrum Match: The sample analyte's mass spectrum must be a statistically significant match (e.g., ≥ 90% similarity) to the reference spectrum in a certified library.
- Quantification: The concentration of the analyte in the sample is calculated by comparing its peak area to the calibration curve.
- Quality Review: A second qualified scientist must independently review the raw data, processing methods, and results to confirm the findings.
- Compound Identification: A positive identification requires two independent data points:
Phase 6: Forensic Reporting
- Objective: To communicate the findings in a clear, accurate, and impartial report that withstands legal scrutiny.
- Protocol: The report must include the sample identifier, a description of the methods used (referencing the validated SOP), the results with appropriate uncertainty measurements, a statement of the findings, and the signatures of the analyzing and reviewing scientists. The expert must be prepared to testify that the analysis followed a reliable application of the principles and methods (per Rule 702(d)) and explain the underlying science to the court.
For the forensic researcher, the journey from a raw sample to a court-ready conclusion is governed by a dual commitment: unwavering scientific integrity and strict legal adherence. The Daubert Standard and Federal Rule of Evidence 702 provide a structured framework for judges to assess the reliability of expert testimony, while the Frye Standard continues to emphasize community consensus in several jurisdictions. For a foundational technique like GC-MS, general acceptance is a given, but admissibility in any specific case hinges on the demonstrably reliable application of the method—from an unbroken chain of custody and rigorous sample preparation to robust instrument calibration and unbiased data interpretation. By embedding the principles of Daubert and Rule 702 directly into their experimental protocols, forensic scientists and drug development professionals ensure that their work is not only scientifically sound but also forensically defensible, thereby faithfully serving the intertwined causes of science and justice.
Gas chromatography-mass spectrometry (GC-MS) has long been a cornerstone technique in forensic research, providing the separation power and reproducible mass spectra necessary for identifying unknown compounds in complex evidentiary samples [94]. Despite its established role, conventional one-dimensional GC-MS faces significant challenges in separating highly complex mixtures due to limited peak capacity, often resulting in co-elution of compounds and difficult-to-deconvolute mass spectra [95] [96]. Comprehensive two-dimensional gas chromatography-mass spectrometry (GC×GC-MS) has emerged as a powerful advancement that addresses these limitations through orthogonal separation mechanisms, dramatically enhancing resolution and compound identification capabilities [95] [97].
This technical analysis provides a comparative examination of GC-MS and GC×GC-MS technologies within the context of forensic research, with particular emphasis on quantitative metrics of peak capacity, metabolite coverage, and practical implications for biomarker discovery in complex biological matrices. The fundamental distinction between these platforms lies in their separation approach: while GC-MS employs a single separation column, GC×GC-MS utilizes two separate columns with different stationary phases connected via a thermal modulator, creating an orthogonal separation system where compounds that co-elute from the first dimension may be separated in the second dimension [95] [97].
Fundamental Principles and Instrumentation
GC-MS Instrumentation and Operating Principles
The GC-MS platform consists of a gas chromatograph interfaced with a mass spectrometer through a transfer line [97]. Samples are injected into the GC inlet, vaporized, and carried by an inert gas (typically helium) through a capillary column (commonly 30-60 meters in length) containing a stationary phase [94] [97]. Separation occurs based on compound volatility and affinity for the stationary phase, with the oven temperature precisely programmed to facilitate elution of different compounds at different times [95]. Eluting compounds then enter the mass spectrometer, where they are ionized (typically by electron ionization at 70 eV), mass-analyzed, and detected [98] [94].
The mass spectra generated by electron ionization are characterized by reproducible fragmentation patterns that enable library matching against extensive commercial databases such as the NIST Mass Spectral Library, making GC-MS particularly valuable for identifying unknown compounds in forensic applications [98] [99]. Common mass analyzer configurations include quadrupole, time-of-flight (TOF), and ion trap systems, each offering different trade-offs in scan speed, resolution, and mass accuracy [100] [97].
GC×GC-MS Instrumentation and Orthogonal Separation
GC×GC-MS builds upon the fundamental GC-MS platform by incorporating a second dimension of separation [95] [97]. The system employs two serially connected columns with different stationary phase chemistries (e.g., a non-polar primary column and a moderately polar secondary column) interfaced through a thermal modulator [95]. The modulator periodically traps, focuses, and reinjects effluent from the first dimension onto the second dimension column, which is typically much shorter (1-2 meters) and operated at a higher temperature than the primary column [95].
This two-dimensional separation mechanism provides a multiplicative increase in peak capacity, as compounds that co-elute in the first dimension may be separated based on different chemical properties (such as polarity) in the second dimension [95] [97]. The result is significantly enhanced resolution of complex mixtures, which is particularly valuable in forensic analysis where evidentiary samples often contain numerous compounds at varying concentration levels.
Figure 1: Instrumental workflow comparison between GC-MS and GC×GC-MS platforms
Comparative Technical Performance
Peak Capacity and Separation Efficiency
Peak capacity, defined as the maximum number of chromatographic peaks that can be separated within a given analysis time, represents a fundamental differentiator between GC-MS and GC×GC-MS systems [95]. In conventional GC-MS, peak capacity is limited by the length, diameter, and stationary phase chemistry of a single column, with practical limits typically ranging from several hundred to approximately 1,000 theoretical plates under optimized conditions [95]. In contrast, GC×GC-MS provides a theoretical peak capacity equivalent to the product of the peak capacities of each dimension, typically reaching 10,000 or more theoretical plates [95] [97].
This dramatic increase in separation power directly addresses one of the most significant challenges in forensic analysis: co-elution of compounds in complex mixtures. In a direct comparison study analyzing human serum samples, the GC×GC-MS platform detected approximately three times as many peaks as the GC-MS platform at a signal-to-noise ratio ≥ 50 [95]. Manual verification confirmed that this improvement resulted primarily from the superior resolution of chromatographic peaks, minimizing peak overlap and facilitating more accurate spectrum deconvolution for compound identification and quantification [95].
Metabolite Coverage and Identification Confidence
The enhanced separation capability of GC×GC-MS directly translates to improved metabolite coverage and identification confidence in complex samples [95] [97]. In a comparative study of 109 human serum samples, three times the number of metabolites were successfully identified by mass spectrum matching (with a spectral similarity score Rsim ≥ 600) using GC×GC-MS compared to conventional GC-MS [95]. This improvement is particularly significant for low-abundance metabolites and structural isomers that may be obscured by more abundant compounds in one-dimensional separation.
The non-selective nature of GC×GC-MS profiling provides an additional advantage for discovery-based forensic applications, as a single scanned run contains data on every detectable compound in a sample, offering an integrated picture of the sample's chemical composition [96]. This comprehensive approach enables the detection of novel or unexpected compounds that might be missed by targeted analytical methods, making it particularly valuable for forensic toxicology and controlled substance analysis [96].
Table 1: Quantitative Comparison of GC-MS and GC×GC-MS Performance in Metabolite Analysis
| Performance Metric | GC-MS | GC×GC-MS | Improvement Factor |
|---|---|---|---|
| Detected Peaks (SNR ≥ 50) | Baseline | ~3× more peaks | 3× [95] |
| Identified Metabolites (Rsim ≥ 600) | Baseline | ~3× more metabolites | 3× [95] |
| Statistically Significant Biomarkers | 23 metabolites | 34 metabolites | 1.5× [95] |
| Common Biomarkers Detected | 9 metabolites | 9 metabolites | Equivalent [95] |
| Separation Mechanism | Single dimension | Orthogonal two-dimensional | Multiplicative peak capacity [95] [97] |
Sensitivity and Detection Limits
The thermal modulation process in GC×GC-MS provides not only enhanced separation but also improved sensitivity through peak focusing effects [95] [97]. As effluent from the first dimension is trapped, focused, and released as narrow bands onto the second dimension column, analyte concentrations are increased, leading to higher signal-to-noise ratios and lower detection limits [95]. This sensitivity enhancement is particularly beneficial in forensic applications where analyte concentrations may be low, such as in the detection of drug metabolites, trace evidence analysis, or environmental contaminant monitoring.
It is noteworthy that the sensitivity advantages of GC×GC-MS may necessitate operational adjustments; for example, one comparative study reported using a 30:1 split ratio for GC×GC-MS analysis compared to splitless injection for GC-MS "because of increased chromatographic peak heights due to modulator peak focusing" [95].
Experimental Methodology for Comparative Analysis
Sample Preparation and Derivatization Protocols
For comprehensive metabolomic analysis using either GC-MS or GC×GC-MS platforms, proper sample preparation and chemical derivatization are critical steps that enable the analysis of non-volatile or thermally labile compounds [95] [98]. The following optimized protocol has been employed in comparative studies of human serum samples:
Protein Precipitation and Metabolite Extraction: Add 100 µL of serum to 1 mL of ice-cold extraction solvent (methanol/chloroform, 3:1 v:v) containing internal standards (e.g., 10 µg/mL heptadecanoic acid and 10 µg/mL norleucine) [95]. Vortex briefly and centrifuge for 15 minutes at 18,000 rcf at 4°C [95]. Transfer supernatant to a clean vial and dry under a gentle stream of nitrogen at room temperature [95].
Two-Step Chemical Derivatization:
- Methoximation: Add 50 µL of freshly prepared methoxyamine in pyridine (20 mg/mL) to the dried sample and incubate for 90 minutes at 30°C with continuous shaking at 1400 rpm [95]. This step protects carbonyl groups and reduces ring formation in sugars.
- Silylation: Add 50 µL of N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% trimethylchlorosilane (TMCS) and incubate for 60 minutes at 70°C with shaking at 1400 rpm [95]. This step replaces active hydrogens with trimethylsilyl groups, increasing volatility and thermal stability.
Quality Control Measures: Prepare a pooled quality control (QC) sample from aliquots of all experimental samples and analyze this QC after every 9-10 biological samples to monitor instrument performance and correct for analytical variations [95].
Instrumental Configuration for Comparative Studies
Direct comparison of GC-MS and GC×GC-MS performance requires careful optimization of both platforms to ensure fair evaluation. The following instrumental parameters have been employed in validated comparative studies [95]:
Table 2: Instrumental Parameters for GC-MS and GC×GC-MS Analysis
| Parameter | GC-MS Configuration | GC×GC-MS Configuration |
|---|---|---|
| GC System | Agilent 7890A | Agilent 7890A |
| Mass Spectrometer | LECO Pegasus TOF-MS | LECO Pegasus TOF-MS |
| Primary Column | DB-5 ms UI (60 m × 0.25 mm × 0.25 µm) | DB-5 ms UI (60 m × 0.25 mm × 0.25 µm) |
| Secondary Column | Not applicable | DB-17 ms (1 m × 0.25 mm × 0.25 µm) |
| Injection Mode | Splitless | Split (30:1) |
| Carrier Gas | Helium at 1.0 mL/min | Helium at 1.0 mL/min |
| Oven Program | 60°C for 1 min, then 5°C/min to 300°C, hold 12 min | 60°C for 1 min, then 5°C/min to 300°C, hold 12 min |
| Modulator Parameters | Not applicable | Modulator period: 2.5 s; Temperature offset: +20°C |
| MS Acquisition Rate | 20 spectra/second | 200 spectra/second |
| Mass Range | m/z 45-1000 | m/z 45-1000 |
Data Processing and Statistical Analysis
The complex data generated by both platforms, particularly the three-dimensional data from GC×GC-MS (retention time 1 × retention time 2 × mass spectra), requires specialized processing approaches [95] [99]:
Peak Detection and Deconvolution: Raw instrument data are initially processed using vendor software (e.g., LECO ChromaTOF) to detect peaks and perform spectrum deconvolution [95]. Parameters should be optimized to minimize false positives while maintaining sensitivity for low-abundance compounds.
Peak Alignment and Metabolite Identification: Use specialized algorithms (e.g., DISCO for peak alignment and iMatch for retention index matching) to align peaks across multiple samples [95]. Match mass spectra against reference libraries (NIST, Fiehn, or in-house databases) with appropriate similarity thresholds (e.g., Rsim ≥ 600) [95].
Multivariate Statistical Analysis: Apply principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) to identify metabolites that show significant abundance changes between sample groups [95]. In comparative studies, these analyses revealed 23 significant biomarkers in GC-MS data versus 34 significant biomarkers in GC×GC-MS data from the same sample set [95].
Figure 2: Experimental workflow for GC-MS and GC×GC-MS metabolomics studies
Essential Research Reagent Solutions
Successful implementation of GC-MS or GC×GC-MS analyses requires specific reagents and materials optimized for metabolomic applications. The following table details key research reagent solutions and their functions in the analytical workflow.
Table 3: Essential Research Reagents for GC-MS and GC×GC-MS Metabolomics
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Methoxyamine hydrochloride | Protects carbonyl groups (ketones, aldehydes) by forming methoximes, preventing sugar ring formation and improving chromatography [95] [98] | Prepare fresh in anhydrous pyridine (20 mg/mL); critical for reducing multiple peaks for single compounds [95] |
| N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) | Silylation reagent that replaces active hydrogens with trimethylsilyl groups, increasing volatility and thermal stability [95] [98] | Use with 1% TMCS as catalyst; derivatives -OH, -COOH, -NH, and -SH groups [95] |
| Retention Index Markers (Alkane standard) | Provides reference points for retention time normalization across samples and between instruments [95] | Analyze C10-C40 alkanes at beginning, middle, and end of sample sequence; enables retention index calculation [95] |
| Deuterated Internal Standards | Corrects for variations in sample preparation, injection, and instrument response [101] | Use compound-specific deuterated analogs (e.g., d₃-norleucine, d₃-heptadecanoic acid) for quantification [95] |
| DB-5 ms UI GC Column | Primary separation column (5%-phenyl)-methylpolysiloxane stationary phase provides excellent separation for diverse metabolite classes [95] | Standard 60 m × 0.25 mm × 0.25 µm dimensions provide optimal balance of resolution and analysis time [95] |
| DB-17 ms GC Column | Secondary separation column for GC×GC; (50%-phenyl)-methylpolysiloxane provides orthogonal separation mechanism [95] | Shorter dimensions (1-2 m × 0.25 mm × 0.25 µm) enable rapid second-dimension separation [95] |
Forensic Applications and Implications
The enhanced separation power and metabolite coverage of GC×GC-MS offers significant advantages for forensic applications involving complex mixture analysis. In forensic toxicology, the ability to resolve drug metabolites from endogenous compounds and co-ingested substances improves confidence in identification and quantification [96]. For controlled substance analysis, GC×GC-MS can separate cutting agents, impurities, and manufacturing byproducts that may provide investigative leads through chemical profiling [96] [94].
The non-selective nature of GC×GC-MS data acquisition makes it particularly valuable for suspect screening and retrospective analysis, as full-scan data contains information on all detectable compounds in a sample [96]. This capability enables re-examination of data as new intelligence emerges, without requiring re-analysis of evidentiary samples. Furthermore, the improved sensitivity of GC×GC-MS facilitates the detection of low-abundance compounds that may be forensically significant but obscured in conventional GC-MS analysis [95] [97].
Despite these advantages, GC×GC-MS implementation in forensic laboratories must consider the increased methodological complexity, longer data processing times, and need for specialized expertise in data interpretation [95]. For routine analysis of relatively simple mixtures, conventional GC-MS may provide sufficient resolution with faster turnaround times and simpler data management. However, for the most challenging forensic samples containing hundreds or thousands of chemical components, GC×GC-MS represents a powerful tool that can extract information not accessible through conventional separation techniques.
GC×GC-MS represents a significant advancement over conventional GC-MS in peak capacity, metabolite coverage, and sensitivity for complex mixture analysis. Quantitative comparisons demonstrate approximately three-fold improvements in both peak detection and metabolite identification using GC×GC-MS, with particular advantages for resolving co-eluting compounds and detecting low-abundance metabolites [95]. These capabilities make GC×GC-MS exceptionally valuable for forensic applications involving complex biological samples, environmental analyses, and controlled substance profiling where comprehensive compound detection and confident identification are paramount.
While GC-MS remains a robust and widely accessible platform for many analytical scenarios, GC×GC-MS provides unparalleled separation power for the most challenging samples. The choice between these platforms should be guided by sample complexity, analytical requirements, and available resources. As forensic science continues to confront increasingly sophisticated analytical challenges, GC×GC-MS offers a powerful tool for advancing chemical analysis capabilities in research and casework applications. Future developments in instrumentation, data processing automation, and spectral libraries will likely expand the practical implementation of GC×GC-MS in forensic laboratories, further enhancing its contribution to analytical science and justice system applications.
Gas Chromatography Tandem Mass Spectrometry (GC-MS/MS) represents a significant evolution in analytical technology, addressing critical limitations of traditional single-stage GC-MS. While conventional GC-MS is a powerful tool, it can struggle to provide reliable quantitative and qualitative information when sample matrices are complex and target analytes are at trace concentrations, or when chromatographic and mass resolution is insufficient to separate interfering compounds, such as isomers [102]. The core strength of GC-MS/MS lies in its ability to provide a higher degree of selectivity, specificity, and sensitivity, while also yielding additional unique mass and structural information about target analytes [102].
In a GC-MS/MS system, two mass analyzers are connected in series with a collision cell situated between them. This configuration allows ions separated in the first mass analyzer (MS1) to undergo controlled fragmentation in the collision cell, with the resulting product ions then being separated in the second mass analyzer (MS2) before detection [102]. This two-stage mass analysis process effectively reduces matrix interferences and background noise, delivering superior performance for challenging applications in fields like forensic science, pharmaceutical development, and environmental analysis [103] [104] [105].
Fundamental Principles and Instrumentation of GC-MS/MS
Core Components and Operational Workflow
A GC-MS/MS system integrates a gas chromatograph with a tandem mass spectrometer. The sample mixture is first separated by the GC based on the differential partitioning of analytes between a mobile gas phase and a stationary phase within the column [5] [104]. Upon exiting the analytical column, the separated neutral molecules are ionized, most commonly by Electron Ionization (EI). In EI, electrons produced by a filament are accelerated at 70 electron volts (eV), causing them to knock electrons out of the analyte molecules to produce molecular ions that are radical cations [5].
The instrumental workflow follows these critical steps:
- Primary Mass Selection (MS1): The first mass analyzer (e.g., a quadrupole) selects precursor ions of a specific mass-to-charge ratio (m/z) from the ionized effluent.
- Collision-Induced Dissociation (CID): The selected precursor ions are directed into a collision cell filled with an inert gas (e.g., Argon or Nitrogen). Collisions between the ions and gas atoms convert kinetic energy into internal energy, causing chemical bonds to break and fragment into product ions [106] [102].
- Secondary Mass Analysis (MS2): The resulting product ions are then separated by the second mass analyzer (e.g., another quadrupole) based on their m/z.
- Detection: The separated product ions are detected, and the signal is recorded by the data system [5].
The following diagram illustrates the logical flow of this process in a triple quadrupole instrument:
Key Scan Modes and Their Applications
The two mass analyzers in a GC-MS/MS system can be operated in different modes, enabling a variety of powerful scan functions that enhance analytical capabilities. The most common operational modes are:
- Product Ion Scan: MS1 is fixed to select a specific precursor ion, while MS2 is scanned to acquire all product ions resulting from its fragmentation. This mode is essential for elucidating chemical structures and for confirming the identity of unknowns [103] [102].
- Precursor Ion Scan: MS1 is scanned over a defined mass range, and MS2 is fixed to monitor a specific product ion. This mode identifies all precursor ions that fragment to produce a common, characteristic product ion, which is useful for screening compound families [102].
- Neutral Loss Scan: Both MS1 and MS2 are scanned simultaneously, but with a constant mass offset between them. This detects all precursors that lose a specific neutral molecule (e.g., H₂O, CO₂), which is valuable for identifying compounds with specific functional groups [102].
- Selected/Multiple Reaction Monitoring (SRM/MRM): Both MS1 and MS2 are fixed at specific m/z values to monitor a defined transition from a precursor to a product ion. This mode offers the ultimate sensitivity and selectivity for quantitative trace analysis, as it significantly reduces chemical noise [103] [102].
Table 1: Comparison of Key Operational Modes in GC-MS/MS
| Scan Mode | MS1 Function | MS2 Function | Primary Application |
|---|---|---|---|
| Product Ion Scan | Fixed on one m/z | Scans a mass range | Structural elucidation, confirmation of identity |
| Precursor Ion Scan | Scans a mass range | Fixed on one m/z | Identifying all precursors of a common fragment |
| Neutral Loss Scan | Scans a mass range | Scans with a fixed mass offset | Detecting losses of specific neutral molecules |
| Multiple Reaction Monitoring (MRM) | Fixed on one m/z | Fixed on one m/z | High-sensitivity quantitation of target compounds |
Analytical Advantages: Specificity and Sensitivity
Enhanced Specificity and Reduced Background Interference
The primary advantage of GC-MS/MS is the dramatic increase in analytical specificity. In complex samples, co-eluting compounds from the matrix can create significant background interference in single-stage GC-MS, leading to false positives or inaccurate quantification. GC-MS/MS overcomes this by adding a second dimension of mass separation.
Specificity is achieved by monitoring a precursor ion → product ion transition that is highly unique to the target analyte. The probability of two different compounds co-eluting from the GC column and sharing the same specific fragmentation pathway is exceedingly low [103]. This makes GC-MS/MS particularly powerful for analyzing target compounds in challenging matrices like biological fluids, food extracts, and environmental samples, where interferences are common. This specific transition acts as a highly selective fingerprint, confirming the analyte's identity with a much higher degree of confidence than a single retention time or a single mass measurement [103].
Improved Sensitivity and Lower Detection Limits
GC-MS/MS also provides exceptional gains in sensitivity, which is critical for detecting trace-level contaminants, metabolites, or biomarkers. This sensitivity boost is largely a result of significant reduction in chemical noise.
In MRM mode, the instrument filters out the vast majority of non-target ions in both the first and second stages of mass analysis. This process drastically lowers the baseline noise. Since the signal from the target analyte remains strong while the background noise is minimized, the signal-to-noise ratio (S/N) increases substantially [103]. This enhanced S/N directly translates to lower limits of detection (LOD), enabling scientists to detect and accurately quantify analytes at concentrations that are challenging or impossible with single-stage GC-MS, sometimes down to femtogram levels [103] [105].
Table 2: Comparative Performance of GC-MS and GC-MS/MS
| Performance Characteristic | GC-MS (Single Quadrupole) | GC-MS/MS (Triple Quadrupole) |
|---|---|---|
| Primary Identification | Retention time & full mass spectrum | Retention time & precursor/product ion transition(s) |
| Quantitation Mode | Selected Ion Monitoring (SIM) | Multiple Reaction Monitoring (MRM) |
| Selectivity | Good | Excellent (reduced matrix interference) |
| Sensitivity (S/N) | Moderate | Very High (due to noise reduction) |
| Structural Information | From full-scan fragmentation pattern | From controlled, multi-stage fragmentation |
| Ideal Use | Unknown identification, less complex samples | Trace analysis in complex matrices, targeted quantitation |
Experimental Protocol: GC-MS/MS Analysis of Δ⁹-THC and Metabolites in Plasma
The following detailed methodology, adapted from a published pharmacokinetic study, illustrates the application of GC-MS/MS for sensitive and specific drug analysis in a biological matrix [105].
Sample Preparation and Derivatization
- Sample Aliquot: Pipette 1 mL of plasma (calibrators, quality controls, and unknown samples) into a clean glass centrifuge tube.
- Internal Standard Addition: Add a known amount of a stable isotope-labeled internal standard (e.g., Δ⁹-THC-d₃) to each tube to correct for variability in extraction and ionization.
- Liquid-Liquid Extraction: Add 5 mL of a n-hexane/ethyl acetate (9:1 v/v) mixture to the sample. Vortex vigorously for 3 minutes and then centrifuge at 3,000 x g for 10 minutes to separate the organic and aqueous layers.
- Solvent Transfer and Evaporation: Transfer the upper organic layer to a new conical tube. Evaporate the extract to dryness under a gentle stream of nitrogen gas in a heated water bath (40°C).
- Derivatization: Reconstitute the dry residue with 50 µL of a derivatization reagent, such as N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA). Cap the tube and heat at 70°C for 30 minutes to form trimethylsilyl (TMS) derivatives, which improve the volatility and chromatographic behavior of the analytes.
Instrumental Configuration and MRM Parameters
- GC Conditions:
- Column: Fused-silica capillary column (e.g., 30 m x 0.25 mm i.d., 0.25 µm film thickness) with a (5%-Phenyl)-methylpolysiloxane stationary phase.
- Inlet Temperature: 280°C, operating in splitless mode.
- Carrier Gas: Helium, constant flow of 1.0 mL/min.
- Oven Program: Initial temperature 150°C (hold 1 min), ramp to 290°C at 20°C/min (hold 5 min).
- MS/MS Conditions:
- Interface Temperature: 280°C.
- Ionization Mode: Electron Ionization (EI) at 70 eV.
- Ion Source Temperature: 230°C.
- Collision Gas: Argon, optimized pressure.
- Data Acquisition: Multiple Reaction Monitoring (MRM) mode. The following transitions are monitored for quantification and confirmation:
- Δ⁹-THC-TMS: Precursor ion m/z 371 → Product ion m/z 289 (Quantifier)
- Δ⁹-THC-TMS: Precursor ion m/z 371 → Product ion m/z 303 (Qualifier)
- Δ⁹-THC-d₃-TMS (Internal Standard): Precursor ion m/z 374 → Product ion m/z 292
Data Analysis and Quantification
- Integration: Integrate the peak areas for the quantifier and qualifier product ions for the analyte and internal standard in all chromatograms.
- Calibration: Construct a calibration curve by plotting the peak area ratio (analyte / internal standard) against the known concentration of the calibrators. A linear regression with 1/x weighting is typically applied.
- Quantification: Calculate the concentration of Δ⁹-THC in unknown samples by interpolating their peak area ratio from the calibration curve.
- Confirmation: Verify the identity of Δ⁹-THC in samples by checking the retention time (within ±0.1 min of the calibrator) and the ratio of the qualifier to quantifier ion areas (within ±20% of the calibrator average).
The workflow for this targeted quantitative analysis is summarized below:
Essential Research Reagents and Materials
Successful GC-MS/MS analysis relies on a suite of high-purity reagents and consumables. The following table details key materials used in the featured experimental protocol and the broader field.
Table 3: Essential Reagents and Materials for GC-MS/MS Analysis
| Item Name | Function / Purpose | Example / Specification |
|---|---|---|
| Derivatization Reagents | Increases volatility and thermal stability of polar analytes; improves chromatographic peak shape. | N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) |
| Internal Standards | Corrects for sample loss during preparation and instrument variability; essential for accurate quantitation. | Stable isotope-labeled analogs (e.g., Δ⁹-THC-d₃), not present in the original sample. |
| High-Purity Solvents | Used for sample extraction, reconstitution, and mobile phase; minimizes background contamination. | HPLC/Grade n-Hexane, Ethyl Acetate, Methanol |
| Inert Gases | Carrier Gas: Transports sample through GC. Collision Gas: Induces fragmentation in CID. | Helium (Carrier), Argon or Nitrogen (Collision Gas) |
| Capillary GC Columns | Separates the complex sample mixture into individual components prior to mass analysis. | Fused silica with (5%-Phenyl)-methylpolysiloxane stationary phase; typical dimensions: 15-30 m x 0.25 mm i.d., 0.25 µm film. |
Advanced Applications in Forensics and Pharmaceutical Research
The unique capabilities of GC-MS/MS make it indispensable in fields requiring the highest levels of certainty and sensitivity.
- Forensic Toxicology and Drug Testing: GC-MS/MS is the gold standard for confirming the presence of drugs of abuse and their metabolites in biological samples. Its high specificity allows it to definitively distinguish target compounds from matrix interferences, which is crucial for legal proceedings. It provides the sensitivity needed to detect low ng/mL concentrations of substances like synthetic opioids, benzodiazepines, and cannabinoids in urine, blood, and hair [105].
- Pharmaceutical Development: In pharmacokinetic studies, GC-MS/MS is used to achieve the ultra-low detection limits required to characterize the concentration-time profile of a drug and its active metabolites in plasma, enabling precise measurement of key parameters like half-life and bioavailability [105].
- Trace Contaminant Analysis: The technique is ideal for monitoring pesticide residues, environmental pollutants, and endogenous metabolites at trace levels within highly complex sample matrices [104].
- Emerging Forensic Applications: Research continues to expand its use. A 2025 study demonstrated the combination of GC-Ion Mobility Spectrometry (GC-IMS) with machine learning to classify the temporal evolution stages of gel-pen ink for authenticating disputed documents, highlighting the ongoing innovation in chromatographic-mass spectrometric techniques for forensic science [107].
Optimizing GC-MS/MS Performance
Method Development and Collision Energy Optimization
A critical step in developing a robust GC-MS/MS method is optimizing the collision energy in the CID cell. The degree of fragmentation is directly controlled by the energy supplied, as different chemical bonds require different activation energies for breakage [102].
Table 4: Collision-Induced Dissociation (CID) Energy Optimization
| Collision Energy Setting | Observed Effect on Precursor Ion | Resulting Mass Spectrum & Application |
|---|---|---|
| Low (e.g., 0-10 V) | Minimal fragmentation; precursor ion remains abundant. | Spectrum dominated by molecular ion; useful for molecular weight confirmation. |
| Medium (e.g., 10-30 V) | Moderate fragmentation; a balance of precursor and product ions. | Provides structural fragments while retaining molecular ion information; good for library matching. |
| High (e.g., >30 V) | Extensive fragmentation; precursor ion is significantly depleted. | Spectrum shows abundant low m/z product ions; ideal for selecting a unique, high-intensity fragment for MRM. |
The process involves performing a product ion scan on the pure target analyte at a range of collision energies. The energy that produces the most abundant and structurally specific product ion is typically chosen for the MRM transition.
Limitations and Considerations
Despite its power, GC-MS/MS has limitations. The most significant is that the sample must be volatile and thermally stable enough to pass through the gas chromatograph without decomposing [104]. For non-volatile or thermally labile compounds, liquid chromatography-tandem mass spectrometry (LC-MS/MS) is often a more suitable alternative. Furthermore, GC-MS/MS instrumentation is more complex and costly to acquire and maintain than single-stage GC-MS. Finally, method development, particularly the optimization of MRM transitions and collision energies, requires significant expertise and time.
Gas chromatography-mass spectrometry (GC-MS) is long established as the "gold standard" in forensic trace evidence analysis due to its versatility in isolating and analyzing different components in unknown mixtures with minimal method development for each new sample [21]. This technique combines the separation power of gas chromatography with the identification capabilities of mass spectrometry. In a typical GC-MS system, molecules are injected into the gas chromatograph, separated on a capillary column as oven temperature increases, and then eluted sequentially into the mass spectrometer's ionization chamber where they are ionized (commonly by electron impact at 70 eV) and fragmented [108]. The resulting fragments are separated by their mass-to-charge ratio, producing a chromatogram that combines retention time data with fragment ion patterns that can be searched against extensive spectral libraries [108].
Despite its powerful capabilities, traditional one-dimensional GC-MS faces significant limitations when analyzing highly complex forensic samples, primarily due to coelution of compounds that overwhelms the separation capacity of a single column [21]. Comprehensive two-dimensional gas chromatography-mass spectrometry (GC×GC-MS) represents a revolutionary advancement that addresses this fundamental limitation. By connecting two separate columns of different stationary phases via a modulator, GC×GC-MS provides two independent separation mechanisms, dramatically increasing the peak capacity and overall resolution of the analysis [7]. The modulator, often called the "heart" of the GC×GC system, preserves separation from the first dimension by capturing narrow retention time windows and injecting them as focused pulses into the secondary column for further separation [7]. This review evaluates the technological and legal readiness of GC×GC-MS for routine implementation in forensic laboratories, examining its applications across diverse evidence types and the pathway to court admissibility.
Technological Fundamentals of GC×GC-MS
System Architecture and Operational Principles
The GC×GC-MS system builds upon traditional GC-MS through several key components and operational modifications that enable its enhanced separation capabilities:
Column Configuration: A primary column (1D) with a specific stationary phase (typically non-polar) is connected in series to a secondary column (2D) with a different stationary phase (typically polar) via a modulator [7]. This setup provides two distinct separation mechanisms—for instance, separation by volatility in the first dimension and by polarity in the second dimension.
Modulator Types: Three commercially available modulator types exist: thermal modulation (TM), Deans switch (DS), and differential flow modulation (DFM) [21]. The modulator's critical function is to collect, focus, and reinject effluent from the first dimension onto the second dimension at precise intervals (typically 1-5 seconds, known as the modulation period) [7].
Detection Advancements: While early GC×GC systems used flame ionization detection (FID) or standard mass spectrometry, current implementations increasingly employ high-resolution time-of-flight mass spectrometry (TOF-MS) and tandem MS systems, which provide the rapid acquisition rates and sensitivity needed to capitalize on the enhanced separation [7].
The separation process follows a systematic pathway: (1) sample introduction onto the primary column; (2) initial separation based on affinity for the first stationary phase; (3) modulation of narrow effluent bands onto the secondary column; (4) rapid secondary separation based on a different chemical property; and (5) detection with specialized MS systems capable of fast data acquisition [7].
Comparative Advantages Over Traditional GC-MS
The two-dimensional separation mechanism of GC×GC-MS provides several distinct analytical advantages critical for forensic applications:
Enhanced Peak Capacity: The theoretical peak capacity equals the product of the peak capacities of each dimension, far exceeding single-dimension separation [7].
Improved Signal-to-Noise Ratio: The focusing effect of modulation concentrates analytes into narrower bands, increasing detectability of trace compounds [7].
Structured Chromatograms: Chemically related compounds form recognizable patterns in the 2D separation space, facilitating identification of unknown components [21].
Deconvolution of Coelutions: Compounds that coelute in the first dimension are often resolved in the second dimension, eliminating a major limitation of traditional GC-MS [21].
Figure 1: GC×GC-MS Analytical Workflow. The process highlights the critical role of the modulator in connecting the two separation dimensions, which provides the enhanced peak capacity central to the technique's forensic value.
Technology Readiness Assessment Across Forensic Applications
The adoption of GC×GC-MS in forensic science must be evaluated through the dual lenses of analytical maturity and legal admissibility. Research applications demonstrate varying levels of technological readiness, from proof-of-concept studies to validated methods approaching routine implementation [7].
Table 1: Technology Readiness Levels (TRL) of GC×GC-MS in Forensic Applications
| Forensic Application | Technology Readiness Level | Key Demonstrations | Remaining Challenges |
|---|---|---|---|
| Illicit Drug Analysis | TRL 3 (Applied Research) | Identification of synthetic cannabinoids and novel psychoactive substances [7] | Standardization, reference databases, validation protocols |
| Fire Debris & Ignitable Liquid Analysis | TRL 4 (Development Complete) | Oil spill tracing, petroleum product classification [7] | Implementation in routine casework, error rate determination |
| Sexual Assault Evidence | TRL 3 (Applied Research) | Lubricant differentiation in absence of DNA [21] | Population studies, transfer and persistence data |
| Trace Evidence (Paint, Polymers) | TRL 3 (Applied Research) | Pyrolysis-GC×GC-MS of automotive paints and tires [21] | Method optimization, inter-laboratory reproducibility |
| Toxicology | TRL 2 (Technology Formulation) | Broad-spectrum drug screening in biological samples [7] | Sample preparation schemes, quantitative validation |
| Decomposition Odor Analysis | TRL 3 (Applied Research) | Volatile organic compound profiling for human remains detection [7] | Standardized collection methods, environmental impact studies |
| Chemical, Biological, Nuclear, Radioactive (CBNR) Forensics | TRL 2 (Technology Formulation) | Chemical warfare agent profiling, environmental signature identification [7] | Safety protocols, validation for regulatory compliance |
Forensic Applications Demonstrating Significant Readiness
Fire Debris and Ignitable Liquid Analysis
The analysis of ignitable liquid residues (ILR) in arson investigations represents one of the most mature applications of GC×GC-MS in forensic science, currently positioned at TRL 4 (Development Complete). Research has demonstrated superior capabilities for oil spill tracing and petroleum product classification compared to traditional GC-MS [7]. The complex nature of petroleum-based ignitable liquids, containing hundreds of hydrocarbons, benefits tremendously from the enhanced separation power of GC×GC-MS, particularly in distinguishing chemically similar samples or analyzing degraded or weathered evidence. The structured chromatograms produced allow for pattern recognition of compound classes (normal alkanes, isoparaffins, aromatics) that facilitates classification according to established standards like ASTM E1618-19 [7].
Sexual Assault Evidence Analysis
The analysis of sexual lubricants presents a compelling application for GC×GC-MS, particularly in sexual assault cases where the perpetrator uses condoms to avoid depositing biological evidence. Traditional DNA analysis fails to provide probative results in approximately 30% of sexual assault kits [21]. GC×GC-MS enables the differentiation of complex, natural oil-based lubricants that contain numerous coeluting compounds in traditional GC-MS. Experimental protocols involve hexane solvent extraction of lubricant residues, followed by GC×GC-MS analysis using a column set typically consisting of a non-polar primary column and a polar secondary column [21]. The resulting two-dimensional chromatograms reveal more than 25 different components in commercial lubricants that show only substantial coelution in traditional GC-MS, providing distinctive chemical fingerprints for comparing evidence from victims, suspects, and crime scenes [21].
Emerging Applications with Demonstration Potential
Paint and Polymer Analysis
Automotive paint evidence from hit-and-run accidents and other vehicle-related crimes presents a complex analytical challenge due to the multilayer nature of paint systems and the hundreds of components present in modern formulations. Current analysis relies on microscopy, infrared spectroscopy, and pyrolysis-GC-MS, with the latter offering the highest discrimination power [21]. Pyrolysis-GC×GC-MS demonstrates significant advantages in resolving coeluting pyrolysis products that limit traditional methods. Experimental protocols employ flash pyrolysis (ramping from 50°C to 750°C at 50°C/s) of small paint chips (~50 µg) followed by comprehensive two-dimensional separation [21]. This approach has successfully resolved critical coelutions such as α-methylstyrene and n-butyl methacrylate in automotive clear coats, which are indistinguishable using single-dimensional separation [21].
Illicit Drug Analysis
The rapidly evolving landscape of illicit drugs, particularly synthetic cannabinoids and novel psychoactive substances, presents increasing challenges for traditional analytical methods. GC×GC-MS shows significant promise for nontargeted forensic applications where a wide range of analytes must be analyzed simultaneously [7]. The technique's increased peak capacity and sensitivity facilitates detection of minor components and metabolites that may be crucial for drug identification and profiling. While research in this area remains primarily at the applied research stage (TRL 3), studies have demonstrated successful characterization of complex drug mixtures that would challenge conventional GC-MS systems [7].
Table 2: Essential Research Reagents and Materials for GC×GC-MS Forensic Analysis
| Reagent/Material | Function in Analysis | Application Examples |
|---|---|---|
| N-trimethylsilyl-N-methyl trifluoroacetamide (MSTFA) | Derivatization of non-volatile compounds for analysis [109] | Amino acid analysis in biological samples |
| Tris(2-carboxyethyl)phosphine (TCEP) | Reduction of disulfide bonds in complex mixtures [109] | Thiol analysis in saliva and other biological fluids |
| Anhydrous Pyridine | Catalyst for derivatization reactions [109] | Preparation of volatile derivatives for GC analysis |
| Hexane | Solvent extraction of non-polar compounds [21] | Lubricant extraction from forensic evidence |
| Acetonitrile (MeCN) | Protein precipitation and sample cleanup [109] | Biological sample deproteinization prior to analysis |
| Quadrupole Mass Spectrometer | Standard detection for GC×GC systems [21] | General forensic screening applications |
| Time-of-Flight (TOF) Mass Spectrometer | High-speed detection for fast GC×GC peaks [7] | Complex mixture analysis with rapid acquisition needs |
| Thermal Modulator | Focusing and transferring effluent between columns [7] | Applications requiring high peak capacity separation |
Legal Admissibility Framework
For any analytical method to transition from research to routine forensic application, it must satisfy stringent legal admissibility standards. The United States court system operates under either the Frye Standard (general acceptance in the relevant scientific community) or the Daubert Standard (which includes testing, peer review, error rates, and acceptance) [7]. The Federal Rule of Evidence 702 codifies these requirements, while Canada employs the Mohan Criteria focusing on relevance, necessity, absence of exclusionary rules, and properly qualified experts [7].
Table 3: Legal Standards for Forensic Method Admissibility
| Legal Standard | Jurisdiction | Key Requirements | Implications for GC×GC-MS |
|---|---|---|---|
| Frye Standard | Some U.S. States | General acceptance in relevant scientific community [7] | Requires broad consensus and peer-reviewed literature |
| Daubert Standard | Federal & Some U.S. States | Testing, peer review, error rates, standards, acceptance [7] | Demands validation studies and error rate quantification |
| Federal Rule of Evidence 702 | U.S. Federal Courts | Sufficient facts/data, reliable principles/methods, proper application [7] | Necessitates rigorous method validation and protocol standardization |
| Mohan Criteria | Canada | Relevance, necessity, no exclusionary rules, qualified expert [7] | Emphasizes fit-for-purpose validation and expert competency |
The pathway to courtroom admissibility for GC×GC-MS requires systematic attention to these legal standards throughout method development and validation. Specific requirements include:
Proof of Reliability: Demonstrated through intra- and inter-laboratory validation studies that establish reproducibility and repeatability under casework conditions [7].
Error Rate Determination: Quantitative assessment of false positive and false negative rates across the method's applicable scope [7].
Standardization: Development and implementation of standardized protocols, quality assurance measures, and data interpretation guidelines [7].
Professional Competency: Establishment of training requirements and certification programs for forensic practitioners using GC×GC-MS technology [7].
Current research indicates that while GC×GC-MS shows tremendous analytical promise across multiple forensic disciplines, significant work remains in addressing these legal requirements before widespread adoption in casework and expert testimony [7].
Experimental Protocols and Methodologies
Sexual Lubricant Analysis by GC×GC-MS
The protocol for analyzing sexual lubricant evidence demonstrates the application of GC×GC-MS to complex forensic mixtures [21]:
Sample Preparation: Lubricant residues are solvent-extracted using hexane. The extract is concentrated under gentle nitrogen flow if necessary.
Instrumental Conditions:
- GC System: 7890B Gas Chromatograph with split/splitless injector
- Primary Column: Non-polar stationary phase (e.g., 100% dimethylpolysiloxane)
- Secondary Column: Polar stationary phase (e.g., 50% phenyl polysiloxane)
- Modulator: Differential flow modulation (DFM) system
- Mass Spectrometer: 5977 Quadrupole MS with electron ionization (70 eV)
- Temperature Program: Oven temperature ramped from 50°C to 280°C at specific rate
Data Analysis: Two-dimensional chromatograms are evaluated for pattern recognition and specific target compounds. Comparison of unknown samples to reference collections enables classification and potential source attribution.
Paint and Tire Analysis by Pyrolysis-GC×GC-MS
The analysis of polymeric materials like automotive paints and tires requires pyrolysis prior to chromatographic separation [21]:
Sample Preparation: Small samples (~50 µg) are collected from evidence and placed in pyrolysis tubes.
Pyrolysis Conditions:
- Instrument: Pyroprobe 4000
- Temperature Program: 50°C for 2 seconds, ramped to 750°C at 50°C/s, held for 2 seconds
- Interface Temperature: Maintained at 300°C to prevent recondensation of pyrolysates
GC×GC-MS Analysis:
- Utilizing the same column configuration and mass spectrometric detection as the lubricant method
- Focus on resolving coeluting pyrolysis products like α-methylstyrene and n-butyl methacrylate
Data Interpretation: Pattern recognition of polymer-specific markers and comparative analysis between questioned and known samples.
Figure 2: Implementation Roadmap for GC×GC-MS in Forensic Laboratories. The pathway highlights the critical stages from current research status to full routine implementation, emphasizing the validation and standardization requirements necessary for legal acceptance.
GC×GC-MS represents a significant analytical advancement with demonstrated potential to enhance forensic science across multiple evidence types. The technology offers proven benefits for complex mixture separation through increased peak capacity, improved sensitivity, and structured chromatograms that facilitate chemical pattern recognition. Current applications in fire debris analysis and sexual assault evidence demonstrate the practical value of this technology in solving real forensic challenges, particularly when traditional methods face limitations.
The pathway to routine implementation requires focused effort on validation, standardization, and error rate determination to meet legal admissibility standards. As research continues to address these requirements and demonstrate practical benefits across diverse evidence types, GC×GC-MS is positioned to transition from specialized research technique to operational forensic tool. The growing body of peer-reviewed literature and increasing research activity indicate strong scientific interest in advancing this technology toward court-ready implementation. Future directions should emphasize collaborative validation studies, development of standardized protocols, and systematic assessment of evidentiary value to fully realize the potential of GC×GC-MS in forensic science.
Conclusion
GC-MS remains an indispensable, robust, and legally recognized tool in the forensic scientist's arsenal, capable of providing definitive identification for a wide array of evidence. Its strength lies in the powerful synergy of chromatographic separation and mass spectral fingerprinting. For the technique to yield actionable, legally defensible results, a rigorous approach encompassing method validation, systematic troubleshooting, and an understanding of admissibility criteria is non-negotiable. Future directions point toward the integration of more powerful separations like GC×GC-MS and GC-MS/MS to unravel even more complex samples, driving the need for standardized validation protocols and continued research to meet the evolving demands of forensic science and the strictures of the legal system.
References
- 1. Introduction to hyphenated techniques and their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658024/]
- 2. What Are Hyphenated Techniques? [https://labioscientific.com/what-are-hyphenated-techniques/]
- 3. Hyphenated Techniques: LC-MS, GC-MS, and ICP-MS [https://www.labmanager.com/hyphenated-techniques-lc-ms-gc-ms-and-icp-ms-34245]
- 4. A Review on GC-MS Hyphenated Technique [https://www.ajpaonline.com/HTML_Papers/Asian%20Journal%20of%20Pharmaceutical%20Analysis__PID__2021-11-4-7.html]
- 5. GC-MS Principle, Instrument and Analyses ... [https://www.technologynetworks.com/analysis/articles/gc-ms-principle-instrument-and-analyses-and-gc-msms-362513]
- 6. Chromatographic Hyphenation Technology [https://www.solutions.bocsci.com/resources/chromatographic-hyphenation-technology.html]
- 7. Are We Ready for It? A Review of Forensic Applications and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12012292/]
- 8. Gas Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/gas-chromatography-mass-spectrometry-gc-ms/gcms-fundamentals?srsltid=AfmBOoqmRMti4xipOluHegE2NdOSDCwStva0qIi43QGP3iXonp5HTcL2]
- 9. Basic Instrumentation of GCMS [https://www.shimadzu.com/an/service-support/technical-support/gas-chromatograph-mass-spectrometry/overview/basic_instrumentation_of_gcms.html]
- 10. Gas Chromatography Mass Spectrometry (GC-MS) ... [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/gas-chromatography-mass-spectrometry-gc-ms-information.html]
- 11. Fundamentals of Benchtop GC–MS Data Analysis and ... [https://www.chromatographyonline.com/view/fundamentals-of-benchtop-gc-ms-data-analysis-and-terminology]
- 12. Application of gas chromatography with tandem mass ... [https://www.sciencedirect.com/science/article/abs/pii/S002196732500617X]
- 13. Basic Principles of Gas Chromatography [https://www.phenomenex.com/knowledge-center/gc-knowledge-center/basic-principles-of-gas-chromatography-separation-and-analysis?srsltid=AfmBOoq9e-uGNfDKi5KpL_hVeInGBqyc1YjggqMpQKfJE2H_LlC3c_By]
- 14. Visualising Analytes in Gas Chromatography by Staining ... [https://www.sciencedirect.com/science/article/pii/S003991402500027X]
- 15. Rapid GC-MS as a Screening Tool for Forensic Fire Debris ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9888146/]
- 16. 3.1: Principles of Gas Chromatography [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/03%3A_Principles_of_Gas_Chromatography/3.01%3A_Principles_of_Gas_Chromatography]
- 17. Scales of Measurement and Presentation of Statistical Data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6206790/]
- 18. Representing Data | Mathematics for the Liberal Arts Graphically [https://courses.lumenlearning.com/wmopen-mathforliberalarts/chapter/introduction-representing-data-graphically/]
- 19. Color Contrast Guidelines for Text & UI Accessibility [https://daily.dev/blog/color-contrast-guidelines-for-text-and-ui-accessibility]
- 20. Understanding Success Criterion 1.4.11: Non-text Contrast [https://www.w3.org/WAI/WCAG21/Understanding/non-text-contrast.html]
- 21. GC×GC–MS for Forensic Analysis [https://www.spectroscopyonline.com/view/gc-gc-ms-forensic-analysis-1]
- 22. 4.2: Quantitative and Qualitative GC and GC-MS [https://chem.libretexts.org/Courses/Duke_University/CHEM_401L%3A_Analytical_Chemistry_Lab/06%3A_Instrument_Facilities_for_CHEM401L/04%3A_Gas_Chromatogram_Mass_Spectrometer_(GCMS)/4.02%3A_Quantitative_and_Qualitative_GC_and_GC-MS]
- 23. Gas Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/gas-chromatography-mass-spectrometry-gc-ms/gcms-fundamentals?srsltid=AfmBOorDNTumFeHydNc37jG5yzBwIP4ZTNB88Ei5KmpaHj2-kHFnA8Fx]
- 24. Mass Spectrometry [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/spectrpy/massspec/masspec1.htm]
- 25. 12.2: Interpreting Mass Spectra [https://chem.libretexts.org/Courses/Athabasca_University/Chemistry_350%3A_Organic_Chemistry_I/12%3A_Structure_Determination-_Mass_Spectrometry_and_Infrared_Spectroscopy/12.02%3A_Interpreting_Mass_Spectra]
- 26. Mass Spectrometry Applications for Toxicology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5282913/]
- 27. Introduction to mass spectrometry data analysis [https://www.aspect-analytics.com/media-blog/introduction-to-mass-spectrometry-data-analysis]
- 28. Basics of Gas Chromatograph-Mass Spectrometry [https://www.shimadzu.com/an/service-support/technical-support/gas-chromatograph-mass-spectrometry.html]
- 29. Gas Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/gas-chromatography-mass-spectrometry-gc-ms/gcms-fundamentals?srsltid=AfmBOophxwazHvjo_M5PTsATjEngaKvIaQSAe-P4W5GkTqPymqZFi85J]
- 30. Total Ion Chromatogram (TIC) [https://www.shimadzu.com/an/service-support/technical-support/analysis-basics/gcms/fundamentals/retention/total_ion.html]
- 31. The Beginner's Guide to Interpreting GC/MS Results [https://www.innovatechlabs.com/newsroom/1841/how-to-interpret-gas-chromatography-mass-spectrometry-results/]
- 32. Qualitative Methods of GC/MS Analysis:Retention Time ... [https://www.shimadzu.com/an/service-support/technical-support/gas-chromatograph-mass-spectrometry/analysis-results/retention/retention.html]
- 33. Interpreting GC-MS Results [https://www.azolifesciences.com/article/Interpreting-GC-MS-Results.aspx]
- 34. Gas chromatography–mass spectrometry [https://en.wikipedia.org/wiki/Gas_chromatography%E2%80%93mass_spectrometry]
- 35. Mass Spectrometry for Forensic Analysis: An Interview with ... [https://www.spectroscopyonline.com/view/mass-spectrometry-for-forensic-analysis-an-interview-with-glen-jackson]
- 36. Mass spectrometry in Forensics (MS) | Research Starters [https://www.ebsco.com/research-starters/chemistry/mass-spectrometry-forensics-ms]
- 37. Forensic Mass Spectrometry: Scientific and Legal Precedents [https://pmc.ncbi.nlm.nih.gov/articles/PMC10326920/]
- 38. Rapid GC-MS method for screening seized drugs in ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1559279/full]
- 39. A Novel and Validated GC-MS/MS Method for the ... [https://www.mdpi.com/1420-3049/30/22/4478]
- 40. Role of Gas Chromatography in Forensics [https://www.phenomenex.com/knowledge-center/gc-knowledge-center/role-gas-chromatography-forensics?srsltid=AfmBOor6EUZlukqKCT6o1xYqTjt6_MZ2sXjGyy9wZh0RCaHoA5AjX0jY]
- 41. Forensic Perspectives on Gas Chromatography [https://www.chromatographyonline.com/view/forensic-perspectives-on-gas-chromatography]
- 42. GC-MS Sample Preparation [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/gas-chromatography-mass-spectrometry-gc-ms-information/gc-ms-sample-preparation.html]
- 43. Nontargeted Screening Using Gas Chromatography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8624013/]
- 44. Rapid GC-MS method for screening seized drugs in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12185466/]
- 45. Validation of a rapid GC–MS method for forensic seized ... [https://www.sciencedirect.com/science/article/abs/pii/S2468170924000614]
- 46. Validation of a Rapid GC-MS Method for Forensic Seized ... [https://www.nist.gov/publications/validation-rapid-gc-ms-method-forensic-seized-drug-screening-applications]
- 47. Gas Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/gas-chromatography-mass-spectrometry-gc-ms/gcms-fundamentals?srsltid=AfmBOorjgjoEXEdkJ7L9cfT3E9CoIn4BFxefbWX-R71zcGlj8IzQcZtD]
- 48. Gas Chromatography Sample Preparation [https://www.organomation.com/gas-chromatography-sample-preparation]
- 49. GC-MS sample preparation and column choice guide [https://scioninstruments.com/us/blog/gc-ms-sample-preparation-and-column-choice-guide/]
- 50. A tutorial on solid-phase analytical derivatization in sample ... [https://www.sciencedirect.com/science/article/pii/S2772391724000446]
- 51. The Identification of Polyester Fibers Dyed with Disperse Dyes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6384617/]
- 52. Application of dye analysis in forensic fibre and textile ... [https://www.sciencedirect.com/science/article/abs/pii/S0379073817302852]
- 53. Improvement of analytical method for three azo dyes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10314154/]
- 54. Simultaneous determination of nineteen azo dyes in water ... [https://www.nature.com/articles/s41598-024-80097-8]
- 55. Direct Analysis of textile dyes from trace fibers by ... [https://www.ojp.gov/library/publications/direct-analysis-textile-dyes-trace-fibers-automated-microfluidics-extraction]
- 56. - GC in Biomedicine MS [https://www.news-medical.net/life-sciences/GC-MS-in-Biomedicine.aspx]
- 57. Chromatography In Forensic Science | Gas & HPLC [https://scioninstruments.com/us/blog/the-use-of-chromatography-in-forensic-science/]
- 58. What is forensic validation and how is it used? [https://www.envistaforensics.com/knowledge-center/insights/articles/forensic-validation-pay-attention-to-the-man-behind-the-curtain/]
- 59. How NIST Resources Can Help Crime Labs Work Faster [https://www.nist.gov/blogs/taking-measure/speeding-wheels-justice-how-nist-resources-can-help-crime-labs-work-faster]
- 60. OSAC Standards Bulletin - January 2025 | NIST [https://www.nist.gov/magazine/osac-standards-bulletin/january-2025]
- 61. Validation of Analytical Methods for Biomarkers Employed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2744124/]
- 62. Troubleshooting Gas Chromatography: Reduced Peak ... [https://www.chromatographyonline.com/view/troubleshooting-gas-chromatography-reduced-peak-size-loss-sensitivity]
- 63. Sensitivity loss in gc-ms [http://www.chromforum.org/viewtopic.php?t=44671]
- 64. Enhancement and validation of a quantitative GC–MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11480513/]
- 65. Gaining Sensitivity in Environmental GC–MS [https://www.chromatographyonline.com/view/gaining-sensitivity-environmental-gc-ms]
- 66. Development of a gas chromatography–mass spectrometry ... [https://www.sciencedirect.com/science/article/pii/S0308814625032637]
- 67. Systematic investigation into matrix effect compensation in ... [https://www.sciencedirect.com/science/article/abs/pii/S003991402500308X]
- 68. Liquid Chromatography in Forensic Science [https://www.chromatographyonline.com/view/liquid-chromatography-in-forensic-science-advances-in-postmortem-analysis-drug-identification-and-cannabis-differentiation]
- 69. Go With the Flow: Thinking About Carrier Gas Flow in GC [https://www.chromatographyonline.com/view/go-flow-thinking-about-carrier-gas-flow-gc]
- 70. Troubleshooting GC Retention-Time, Efficiency, and Peak- ... [https://www.chromatographyonline.com/view/troubleshooting-gc-retention-time-efficiency-and-peak-shape-problems]
- 71. When GC Retention Times Shift: Practical Advice for ... [https://www.sepscience.com/when-gc-retention-times-shift-practical-advice-for-operators-10822]
- 72. Maintenance and Troubleshooting of GC Columns [https://www.phenomenex.com/knowledge-center/gc-knowledge-center/gc-column-troubleshooting-maintenance?srsltid=AfmBOorr7r3gPBFcw6siLu-p7Ss_0dOGooor7PH51HzibxXF6u96LEtN]
- 73. Reduced Peak Size (Loss of Sensitivity) [https://www.chromatographyonline.com/view/lcgc-blog-troubleshooting-gas-chromatography-part-ii-reduced-peak-size-loss-sensitivity]
- 74. Sudden drop of MS signal [http://www.chromforum.org/viewtopic.php?t=7170]
- 75. Troubleshooting Loss of Signal: Where did my peaks go? [https://www.biotage.com/blog/troubleshooting-loss-of-signal-where-did-my-peaks-go]
- 76. Agilent GC-MS Maintenance: Restek's Quick Reference ... [https://www.restek.com/articles/agilent-gc-ms-maintenance-resteks-quick-reference-guide?srsltid=AfmBOoqWanYpiRsQirBf5E_MZa-ZMCYSIAoPvIdCZVj2L03kSJ5hlT2A]
- 77. How to Maintain a Mass : Best Practices Spectrometer [https://www.linkedin.com/advice/0/what-best-ways-maintain-mass-spectrometer]
- 78. Mass Spectrometry Analyzer Care and Repair [https://www.mks.com/n/mass-spectrometry-analyzer-care-and-repair]
- 79. replacement in Detectors – Baspik mass spectrometers [https://baspik.com/en/services/detectors-replacement-in-mass-spectrometers/]
- 80. Ask the Editor: Seven Preventive Maintenance Tips for Gas ... [https://www.chromatographyonline.com/view/ask-editor-seven-preventive-maintenance-tips-gas-chromatography-systems]
- 81. TECH TIP: Best Practices for GC Preventive Maintenance [https://gentechscientific.com/tech-tip-best-practices-for-gc-preventive-maintenance/]
- 82. GCMS Maintenance: Keep Your GCMS Running [https://www.theoverbrookgroup.com/blog/gcms-maintenance-keeping-your-labs-equipment-running]
- 83. Optimizing GC–MS Methods [https://www.chromatographyonline.com/view/optimizing-gc-ms-methods]
- 84. Guide to GC Column Selection and Optimizing Separations [https://www.restek.com/articles/guide-to-gc-column-selection-and-optimizing-separations?srsltid=AfmBOoqot5GdRaxbnGsHxX3Gvxp3fwzdo_gJ96Dip-b-lIfNbr0xqGvC]
- 85. Frye Standard | Wex | US Law | LII / Legal Information Institute [https://www.law.cornell.edu/wex/frye_standard]
- 86. Frye standard [https://en.wikipedia.org/wiki/Frye_standard]
- 87. The Frye Standard | Expert Testimony, Admissibility, History [https://www.expertinstitute.com/resources/insights/admitting-expert-testimony-under-the-frye-standard-the-ultimate-guide/]
- 88. Daubert standard [https://en.wikipedia.org/wiki/Daubert_standard]
- 89. The Daubert Standard | Expert Testimony, Admissibility ... [https://www.expertinstitute.com/resources/insights/the-daubert-standard-a-guide-to-motions-hearings-and-rulings/]
- 90. Law 101: Legal Guide for the Forensic Expert | Daubert and ... [https://nij.ojp.gov/nij-hosted-online-training-courses/law-101-legal-guide-forensic-expert/pretrial/pretrial-rules-evidence/daubert-and-kumho-decisions]
- 91. Rule 702. Testimony by Expert Witnesses - Cornell Law School [https://www.law.cornell.edu/rules/fre/rule_702]
- 92. Federal Rule of Evidence 702 [https://harvardlawreview.org/print/vol-138/federal-rule-of-evidence-702/]
- 93. Gas Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/gas-chromatography-mass-spectrometry-gc-ms/gcms-fundamentals?srsltid=AfmBOoqvcVJ5y1CXBtzZ2seY9StM7rPuYxnJ63G9khDGF6Y5CsWfbP-h]
- 94. sciencedirect.com/topics/materials-science/ gas - chromatography ... [https://www.sciencedirect.com/topics/materials-science/gas-chromatography-mass-spectrometry]
- 95. Comparison of GC-MS and GC×GC-MS in the Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4849136/]
- 96. Gas chromatography/mass spectrometry (GC/MS) remains ... [https://www.sciencedirect.com/science/article/pii/S0960076010002104]
- 97. Gas Chromatography in Metabolomics: Techniques & ... [https://metabolomics.creative-proteomics.com/resource/gas-chromatography-metabolomics-techniques-instruments.htm]
- 98. GC-MS Metabolomics Analysis [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/mass-spectrometry-applications-area/metabolomics-mass-spectrometry/practical-guide-metabolomics/gc-ms-metabolomics-analysis.html]
- 99. Gas chromatography-mass spectrometry (GC-MS) [https://pubmed.ncbi.nlm.nih.gov/21207291/]
- 100. Latest News on Mass Specs – February 2025 [https://www.phenomenex.com/news/news/2025/02/mass-specs?srsltid=AfmBOorB6DG9D7HtMunh5uv1sBtADpWmmzIj5Q8cDk-N9KMdyyAtXk1W]
- 101. Gas chromatography tandem mass spectrometry offers ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5713679/]
- 102. Core principles [https://www.shimadzu.com/an/service-support/technical-support/analysis-basics/fundamental/core_principles.html]
- 103. Flying High with Sensitivity and Selectivity:- GC–MS to ... [https://www.chromatographyonline.com/view/flying-high-with-sensitivity-and-selectivity---gc-ms-to-gc-ms-ms]
- 104. GC-MS/MS | Tandem Mass Spectrometer [https://www.eag.com/techniques/mass-spec/gc-msms/]
- 105. Gas chromatography/tandem Mass Spectrometry ... [https://pubmed.ncbi.nlm.nih.gov/8122294/]
- 106. Tandem Mass Spectrometry - an overview [https://www.sciencedirect.com/topics/materials-science/tandem-mass-spectrometry]
- 107. Researchers Explore Gas Chromatography to Improve ... [https://www.chromatographyonline.com/view/researchers-explore-gas-chromatography-to-improve-forensic-ink-analysis]
- 108. Gas Chromatography Mass Spectrometry - an overview [https://www.sciencedirect.com/topics/food-science/gas-chromatography-mass-spectrometry]
- 109. Gas Chromatography–Mass Spectrometry Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7729597/]